Information regarding preprints will be updated shortly in this section.

Theo S. Yang, Daniel F Jarosz, Roseanna N Zia
Biophysics 2022
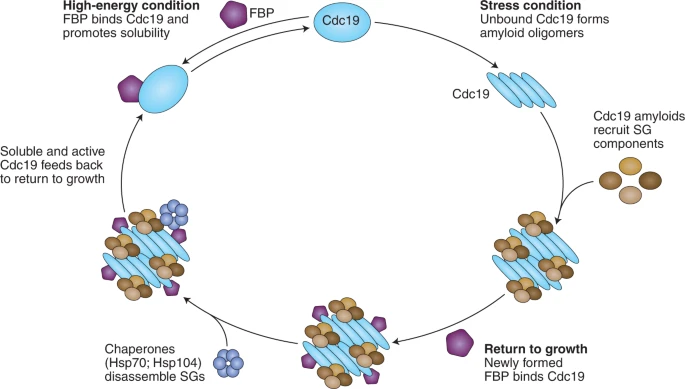
Christopher M Jakobson, Daniel F Jarosz
Nature cell biology 2021
Cells respond to stimuli by reorganizing their contents into subcellular structures. New research demonstrates that yeast pyruvate kinase Cdc19 interacts with fructose-1,6-bisphosphate to coordinate disassembly of stress granules. These findings reveal how proteins can directly sense the cellular energy state to facilitate adaptive reorganization.
Yiwen R Chen, Inbal Ziv, Kavya Swaminathan, Joshua E Elias, Daniel F Jarosz
Biological sciences 2021
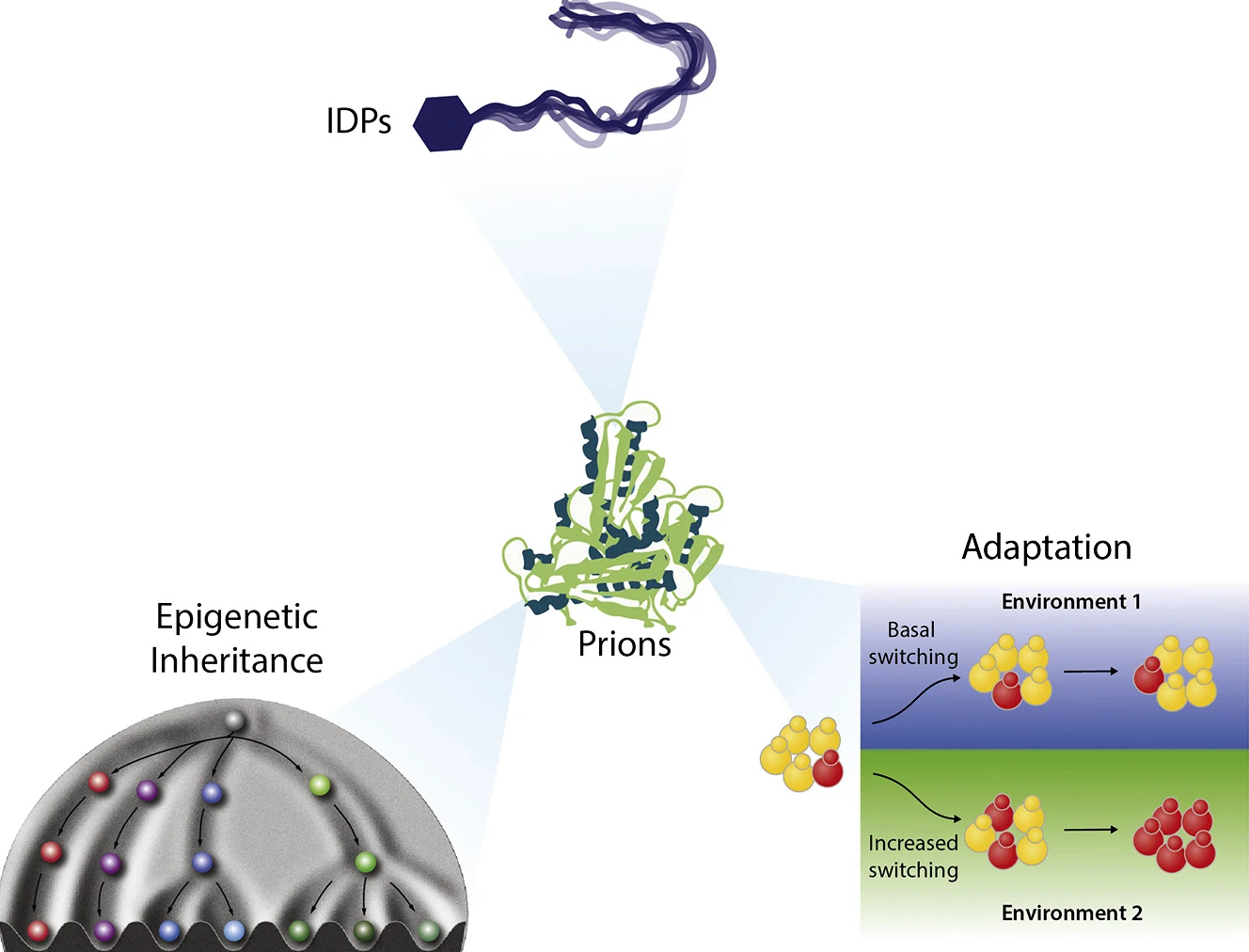
Protein aggregation, particularly in its prion-like form, has long been thought to be detrimental. However, recent studies have identified multiple instances where protein aggregation is important for normal physiological functions. Combining mass spectrometry and cell biological approaches, we developed a strategy for the identification of protein aggregates in cell lysates. We used this approach to characterize prion-based traits in pathogenic strains of the yeast Saccharomyces cerevisiae isolated from immunocompromised human patients. The proteins that we found, including the metabolic enzyme Cdc19, the translation elongation factor Yef3 and the fibrillarin homologue Nop1, are known to assemble under certain physiological conditions. Yet, such assemblies have not been reported to be stable or heritable. Our data suggest that some proteins which aggregate in response to stress have the capacity to acquire diverse assembled states, certain ones of which can be propagated across generations in a form of protein-based epigenetics. This article is part of the theme issue ‘How does epigenetics influence the course of evolution?’
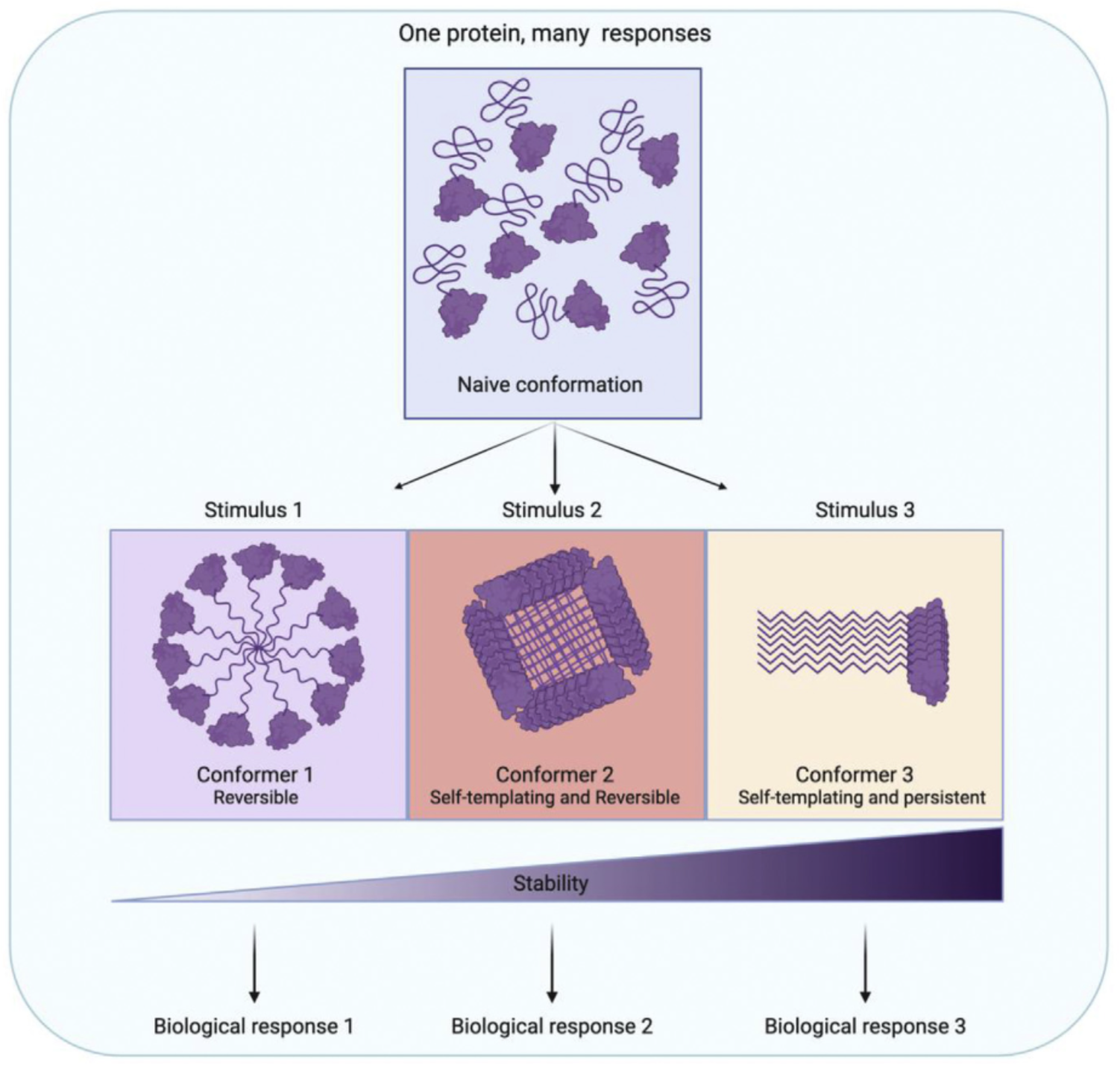
Shady Saad and Daniel F. Jarosz
Current Opinion in Cell Biology. 2021.
Long viewed as paradigm-shifting, but rare, prions have recently been discovered in all domains of life. Protein sequences that can drive this form of self-assembly are strikingly common in eukaryotic proteomes, where they are enriched in proteins involved in information flow and signal transduction. Although prions were thought to be a consequence of random errors in protein folding, recent studies suggest that prion formation can be a controlled process initiated by defined cellular signals. Many are present in normal biological contexts, yet are invisible to most technologies used to interrogate the proteome. Here, we review mechanisms by which protein self-assembly can create a stable record of past stimuli, altering adaptive responses, and how prion behavior is controlled by signaling processes.
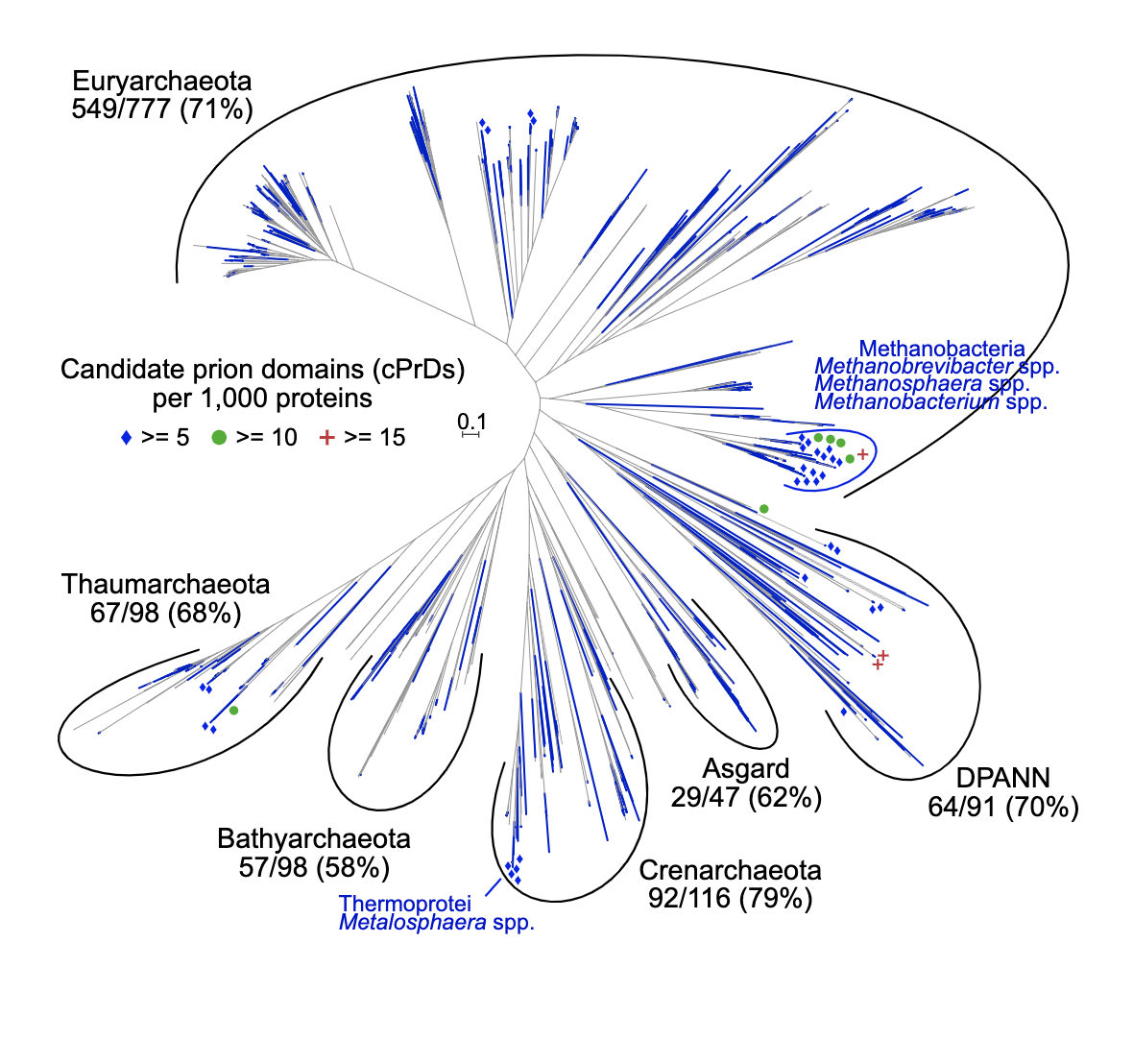
Tomasz Zajkowski, Michael D. Lee, Shamba S. Mondal, Amanda Carbajal, Robert Dec, Patrick D. Brennock, Radoslaw W. Piast, Jessica E. Snyder, Nicholas B. Bense, Wojciech Dzwolak, Daniel F. Jarosz, and Lynn J. Rothschild
Molecular Biology and Evolution. 2021.
Prions, proteins that can convert between structurally and functionally distinct states and serve as non-Mendelian mechanisms of inheritance, were initially discovered and only known in eukaryotes, and consequently considered to likely be a relatively late evolutionary acquisition. However, the recent discovery of prions in bacteria and viruses has intimated a potentially more ancient evolutionary origin. Here we provide evidence that prion-forming domains exist in the domain archaea, the last domain of life left unexplored with regard to prions. We searched for archaeal candidate prion-forming protein sequences computationally, described their taxonomic distribution and phylogeny, and analyzed their associated functional annotations.
Christopher M. Jakobson and Daniel F. Jarosz
Annual Review of Genetics. 2020.
The complexity of heredity has been appreciated for decades: Many traits are controlled not by a single genetic locus but instead by polymorphisms throughout the genome. The importance of complex traits in biology and medicine has motivated diverse approaches to understanding their detailed genetic bases. Here, we focus on recent systematic studies, many in budding yeast, which have revealed that large numbers of all kinds of molecular variation, from noncoding to synonymous variants, can make significant contributions to phenotype. Variants can affect different traits in opposing directions, and their contributions can be modified by both the environment and the epigenetic state of the cell. The integration of prospective (synthesizing and analyzing variants) and retrospective (examining standing variation) approaches promises to reveal how natural selection shapes quantitative traits. Only by comprehensively understanding nature’s genetic tool kit can we predict how phenotypes arise from the complex ensembles of genetic variants in living organisms.
Yiwen R. Chen and Daniel F. Jarosz
Cell Metabolism. 2020.
Covalent cysteine modification by reactive oxygen species (ROS) has been implicated in regulating diverse biological processes, yet global understanding of this modification has remained fragmentary. Developing new approaches for detecting cysteine modification, Xiao et al. (2020) recently charted a comprehensive map of cysteine oxidation across tissues and life stages.
Zachary H. Harvey, Anupam K. Chakravarty, Raymond A. Futia, and Daniel F. Jarosz
Cell. 2020.
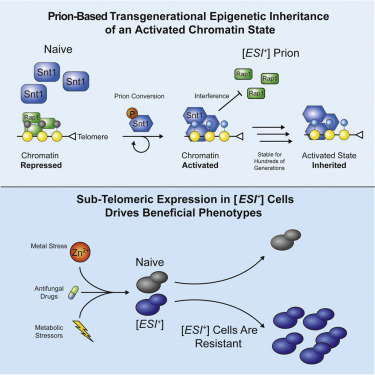
Covalent modifications to histones are essential for development, establishing distinct and functional chromatin domains from a common genetic sequence. Whereas repressed chromatin is robustly inherited, no mechanism that facilitates inheritance of an activated domain has been described. Here, we report that the Set3C histone deacetylase scaffold Snt1 can act as a prion that drives the emergence and transgenerational inheritance of an activated chromatin state. This prion, which we term [ESI+] for expressed sub-telomeric information, is triggered by transient Snt1 phosphorylation upon cell cycle arrest. Once engaged, the prion reshapes the activity of Snt1 and the Set3C complex, recruiting RNA pol II and interfering with Rap1 binding to activate genes in otherwise repressed sub-telomeric domains. This transcriptional state confers broad resistance to environmental stress, including antifungal drugs. Altogether, our results establish a robust means by which a prion can facilitate inheritance of an activated chromatin state to provide adaptive benefit.
Alan K. Itakura, Anupam K. Chakravarty, Christopher M. Jakobson, and Daniel F. Jarosz
Molecular Cell. 2019.
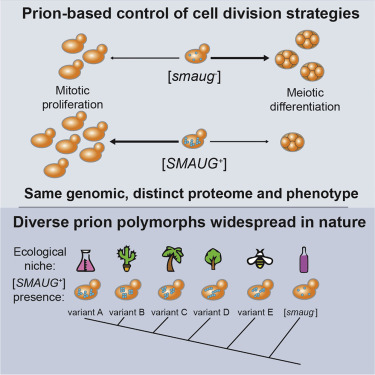
Theory and experiments suggest that organisms would benefit from pre-adaptation to future stressors based on reproducible environmental fluctuations experienced by their ancestors, but the mechanisms driving pre-adaptation remain enigmatic. We report that the [SMAUG+] prion allows yeast to anticipate nutrient repletion after periods of starvation, providing a strong selective advantage. By transforming the landscape of post-transcriptional gene expression, [SMAUG+] regulates the decision between two broad growth and survival strategies: mitotic proliferation or meiotic differentiation into a stress-resistant state. [SMAUG+] is common in laboratory yeast strains, where standard propagation practice produces regular cycles of nutrient scarcity followed by repletion. Distinct [SMAUG+] variants are also widespread in wild yeast isolates from multiple niches, establishing that prion polymorphs can be utilized in natural populations. Our data provide a striking example of how protein-based epigenetic switches, hidden in plain sight, can establish a transgenerational memory that integrates adaptive prediction into developmental decisions.
Anupam K. Chakravarty, Tina Smejkal, Alan K. Itakura, David M. Garcia, and Daniel F. Jarosz
Molecular Cell. 2019.
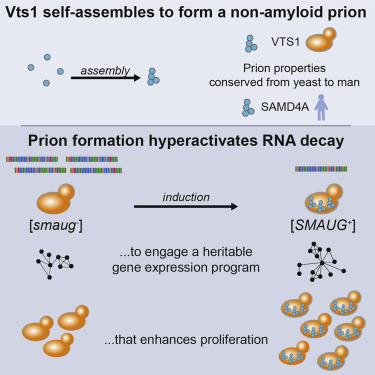
Spatiotemporal gene regulation is often driven by RNA-binding proteins that harbor long intrinsically disordered regions in addition to folded RNA-binding domains. We report that the disordered region of the evolutionarily ancient developmental regulator Vts1/Smaug drives self-assembly into gel-like condensates. These proteinaceous particles are not composed of amyloid, yet they are infectious, allowing them to act as a protein-based epigenetic element: a prion [SMAUG+]. In contrast to many amyloid prions, condensation of Vts1 enhances its function in mRNA decay, and its self-assembly properties are conserved over large evolutionary distances. Yeast cells harboring [SMAUG+] downregulate a coherent network of mRNAs and exhibit improved growth under nutrient limitation. Vts1 condensates formed from purified protein can transform naive cells to acquire [SMAUG+]. Our data establish that non-amyloid self-assembly of RNA-binding proteins can drive a form of epigenetics beyond the chromosome, instilling adaptive gene expression programs that are heritable over long biological timescales.
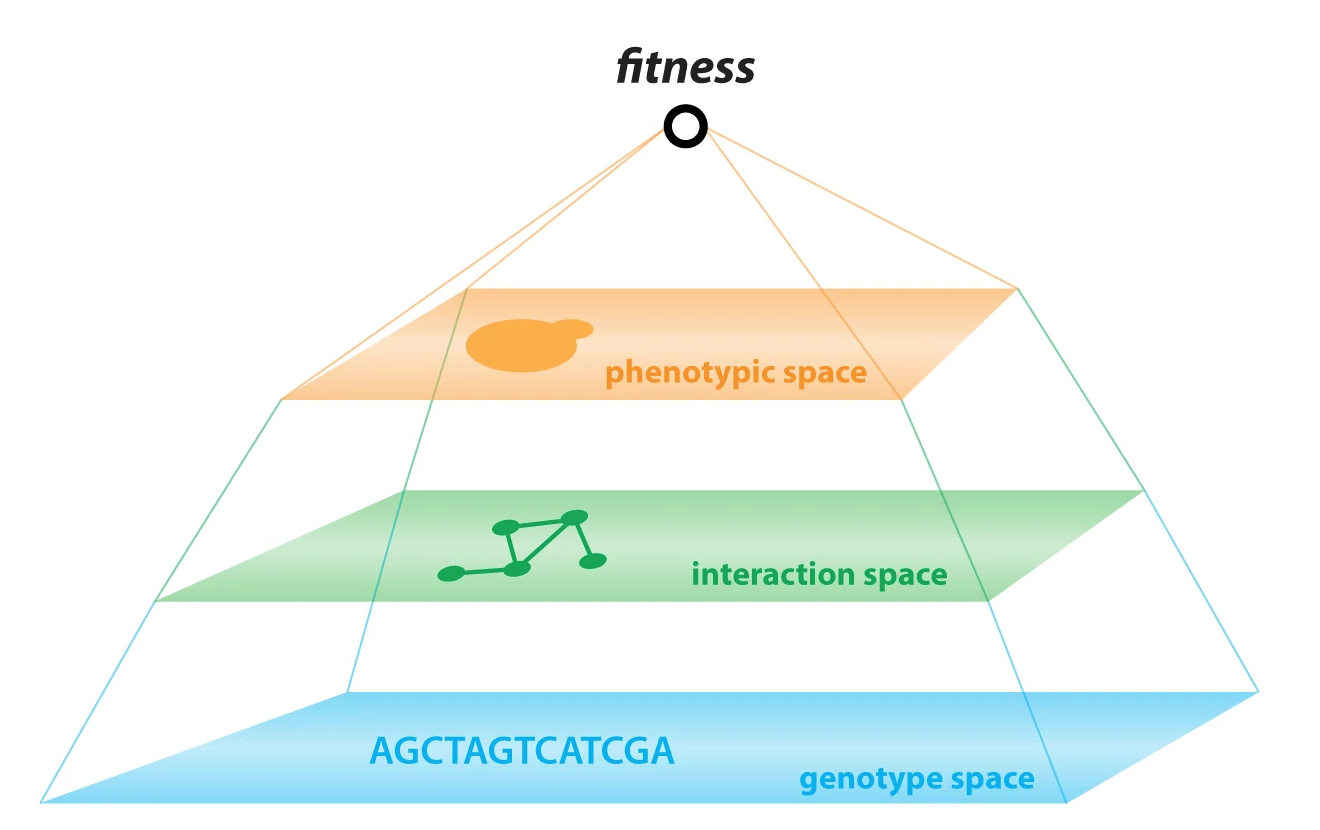
Christopher M. Jakobson and Daniel F. Jarosz.
Cell Systems. 2019.
The statistical complexity of heredity has long been evident, but its molecular origins remain elusive. To investigate, we charted 90 comprehensive genotype-to-phenotype maps in a large population of wild diploid yeast. In contrast to long-standing assumptions, all types of genetic variation contributed similarly to phenotype. Causal synonymous and regulatory variants exhibited distinct molecular signatures, as did nonlinearities in heterozygote fitness that likely contribute to hybrid vigor. Highly pleiotropic variants altered disordered sequences within signaling hubs, and their effects correlated across environments-even when antagonistic-suggesting that large fitness gains bring concomitant costs. Natural genetic networks defined by the causal loci differed from those determined by precise gene deletions or protein-protein interactions. Finally, we found that traits that would appear omnigenic in less powered studies do in fact have finite genetic determinants. Integrating these molecular principles will be crucial as genome reading and writing become routine in research, industry, and medicine.
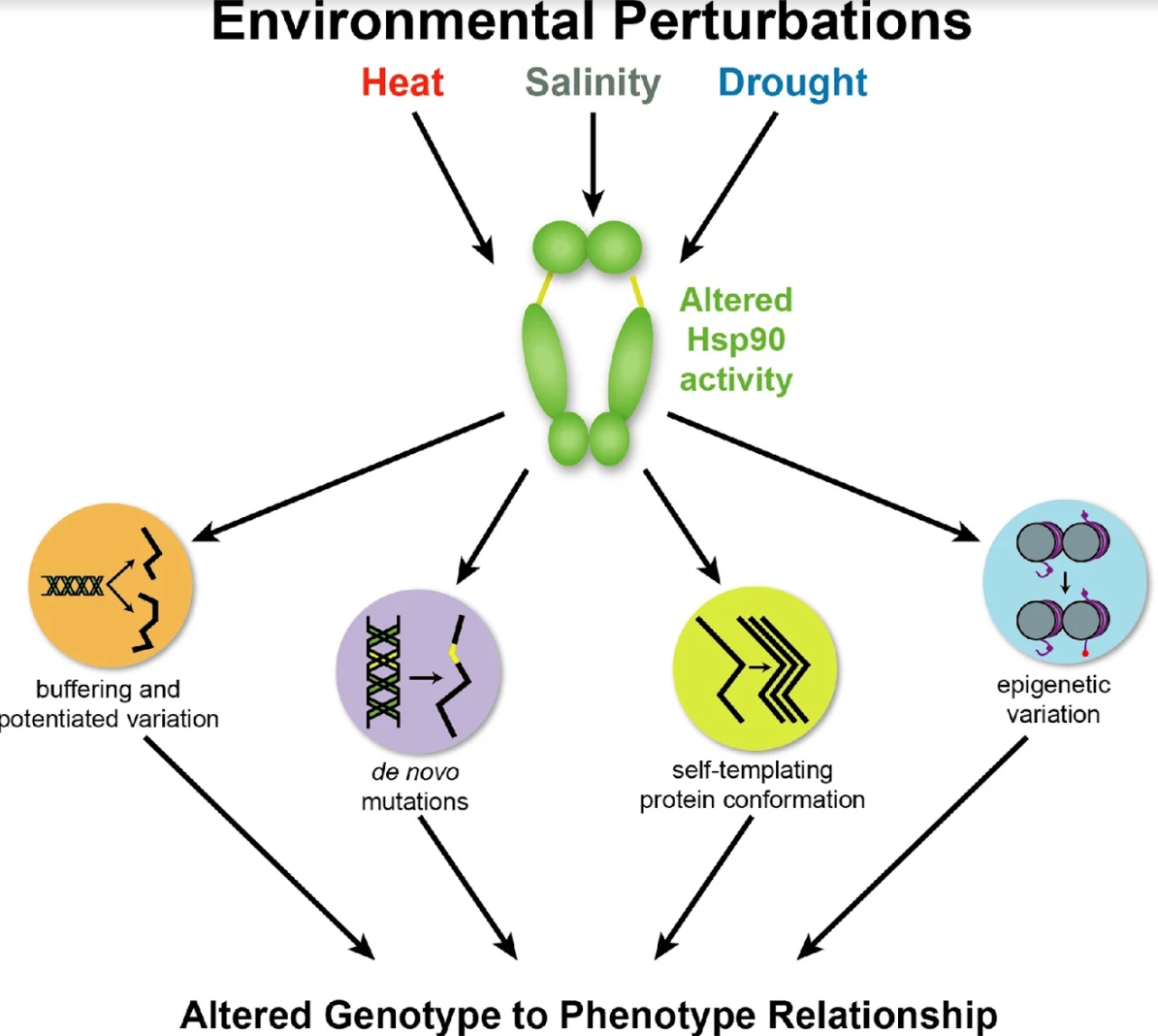
Rebecca A. Zabinsky, Grace A. Mason, Christine Queitsch, and Daniel F. Jarosz.
Seminars in Cell and Developmental Biology. 2019
Canalization, or phenotypic robustness in the face of environmental and genetic perturbation, is an emergent property of living systems. Although this phenomenon has long been recognized, its molecular underpinnings have remained enigmatic until recently. Here, we review the contributions of the molecular chaperone Hsp90, a protein that facilitates the folding of many key regulators of growth and development, to canalization of phenotype – and de-canalization in times of stress – drawing on studies in eukaryotes as diverse as baker’s yeast, mouse ear cress, and blind Mexican cavefish. Hsp90 is a hub of hubs that interacts with many so-called ‘client proteins,’ which affect virtually every aspect of cell signaling and physiology. As Hsp90 facilitates client folding and stability, it can epistatically suppress or enable the expression of genetic variants in its clients and other proteins that acquire client status through mutation. Hsp90’s vast interaction network explains the breadth of its phenotypic reach, including Hsp90-dependent de novo mutations and epigenetic effects on gene regulation. Intrinsic links between environmental stress and Hsp90 function thus endow living systems with phenotypic plasticity in fluctuating environments. As environmental perturbations alter Hsp90 function, they also alter Hsp90’s interaction with its client proteins, thereby re-wiring networks that determine the genotype-to-phenotype map. Ensuing de-canalization of phenotype creates phenotypic diversity that is not simply stochastic, but often has an underlying genetic basis. Thus, extreme phenotypes can be selected, and assimilated so that they no longer require environmental stress to manifest. In addition to acting on standing genetic variation, Hsp90 perturbation has also been linked to increased frequency of de novo variation and several epigenetic phenomena, all with the potential to generate heritable phenotypic change. Here, we aim to clarify and discuss the multiple means by which Hsp90 can affect phenotype and possibly evolutionary change, and identify their underlying common feature: at its core, Hsp90 interacts epistatically through its chaperone function with many other genes and their gene products. Its influence on phenotypic diversification is thus not magic but rather a fundamental property of genetics.
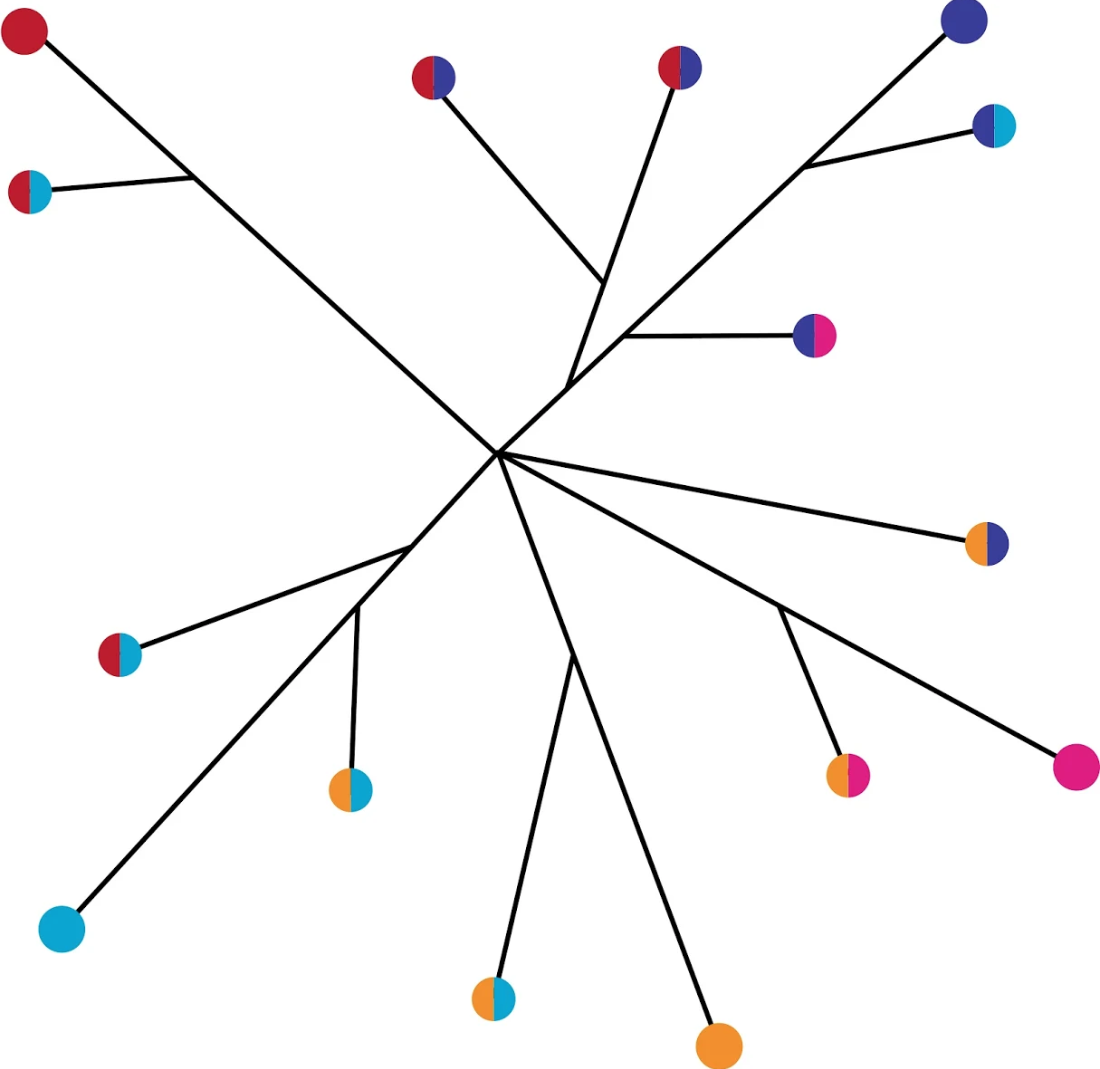
Christopher M. Jakobson, Richard She, and Daniel F. Jarosz.
Nature Communications. 2019
Quantitative genetics aims to map genotype to phenotype, often with the goal of understanding how organisms evolved. However, it remains unclear whether the genetic variants identified are exemplary of evolution. Here we analyzed progeny of two wild Saccharomyces cerevisiae isolates to identify 195 loci underlying complex metabolic traits, resolving 107 to single polymorphisms with diverse molecular mechanisms. More than 20% of causal variants exhibited patterns of emergence inconsistent with neutrality. Moreover, contrary to drift-centric expectation, variation in diverse wild yeast isolates broadly exhibited this property: over 30% of shared natural variants exhibited phylogenetic signatures suggesting that they are not neutral. This pattern is likely attributable to both homoplasy and balancing selection on ancestral polymorphism. Variants that emerged repeatedly were more likely to have done so in isolates from the same ecological niche. Our results underscore the power of super-resolution mapping of ecologically relevant traits in understanding adaptation and evolution.
Lucy J. Xie and Daniel F. Jarosz.
DNA Repair. 2018
From bacteria to humans, ancient stress responses enable organisms to contend with damage to both the genome and the proteome. These pathways have long been viewed as fundamentally separate responses. Yet recent discoveries from multiple fields have revealed surprising links between the two. Many DNA-damaging agents also target proteins, and mutagenesis induced by DNA damage produces variant proteins that are prone to misfolding, degradation, and aggregation. Likewise, recent studies have observed pervasive engagement of a p53-mediated response, and other factors linked to maintenance of genomic integrity, in response to misfolded protein stress. Perhaps most remarkably, protein aggregation and self-assembly has now been observed in multiple proteins that regulate the DNA damage response. The importance of these connections is highlighted by disease models of both cancer and neurodegeneration, in which compromised DNA repair machinery leads to profound defects in protein quality control, and vice versa.
Anupam K. Chakravarty and Daniel F. Jarosz.
Journal of Molecular Biology. 2018
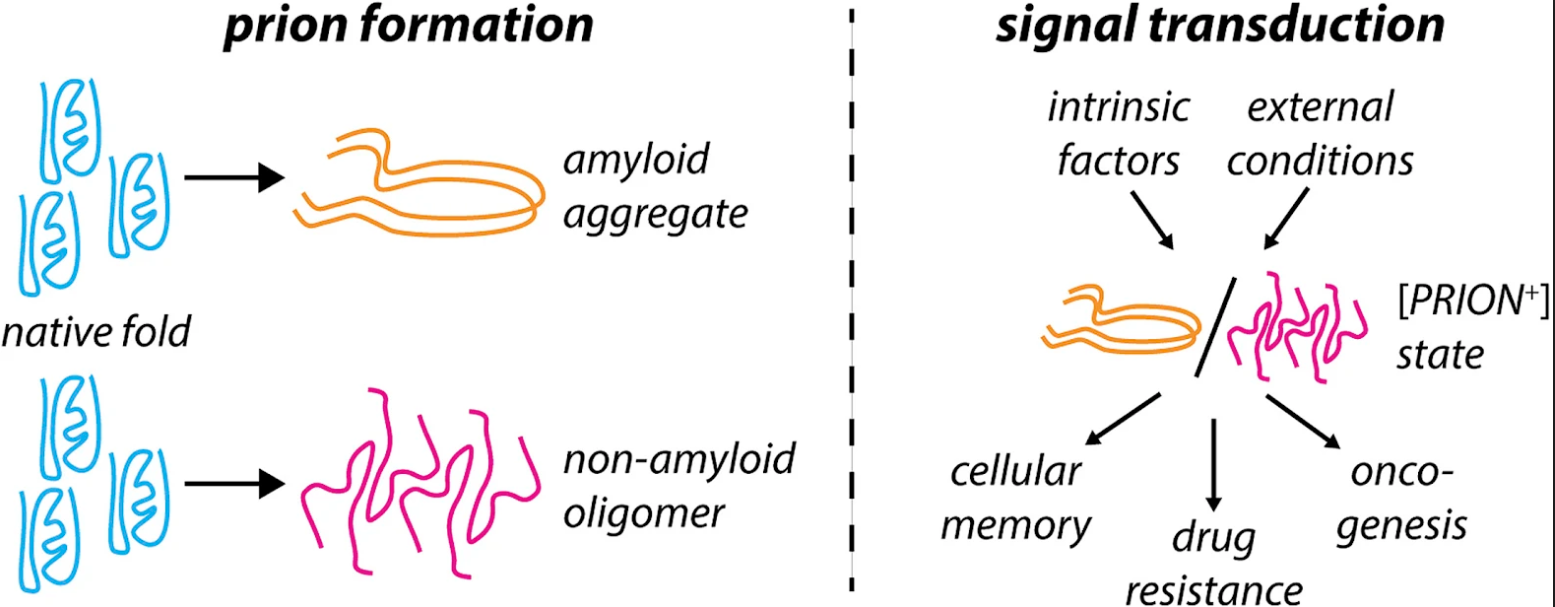
A central tenet of molecular biology is that heritable information is stored in nucleic acids. However, this paradigm has been overturned by a group of proteins called “prions.” Prion proteins, many of which are intrinsically disordered, can adopt multiple conformations, at least one of which has the capacity to self-template. This unusual folding landscape drives a form of extreme epigenetic inheritance that can be stable through both mitotic and meiotic cell divisions. Although the first prion discovered-mammalian PrP-is the causative agent of debilitating neuropathies, many additional prions have now been identified that are not obviously detrimental and can even be adaptive. Intrinsically disordered regions, which endow proteins with the bulk property of “phase-separation,” can also be drivers of prion formation. Indeed, many protein domains that promote phase separation have been described as prion-like. In this review, we describe how prions lie at the crossroads of phase separation, epigenetic inheritance, and evolutionary adaptation.
Christopher M. Jakobson and Daniel F. Jarosz.
Current Opinion in Systems Biology. 2018
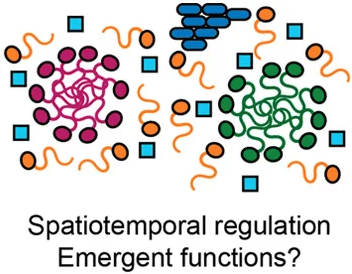
Prion-like proteins have the capacity to adopt multiple stable conformations, at least one of which can recruit proteins from the native conformation into the alternative fold. Although classically associated with disease, prion-like assembly has recently been proposed to organize a range of normal biochemical processes in space and time. Organisms from bacteria to mammals use prion-like mechanisms to (re)organize their proteome in response to intracellular and extracellular stimuli. Prion-like behavior is an economical means to control biochemistry and gene regulation at the systems level, and prions can act as protein-based genes to facilitate quasi-Lamarckian inheritance of induced traits. These mechanisms allow individual cells to express distinct heritable traits using the same complement of polypeptides. Understanding and controlling prion-like behavior is therefore a promising strategy to combat diverse pathologies and organize engineered biological systems.
Alan K. Itakura*, Raymond A. Futia*, and Daniel F. Jarosz.
Biochemistry. 2018
To survive, organisms must orchestrate competing biochemical and regulatory processes in time and space. Recent work has suggested that the underlying chemical properties of some biomolecules allow them to self-organize and that life may have exploited this property to organize biochemistry in space and time. Such phase separation is ubiquitous, particularly among the many regulatory proteins that harbor prion-like intrinsically disordered domains. And yet, despite evident regulation by post-translational modification and myriad other stimuli, the biological significance of many phase-separated compartments remains uncertain. Many potential functions have been proposed, but far fewer have been demonstrated. A burgeoning subfield at the intersection of cell biology and polymer physics has defined the biophysical underpinnings that govern the genesis and stability of these particles. The picture is complex: many assemblies are composed of multiple proteins that each have the capacity to phase separate. Here, we briefly discuss this foundational work and survey recent efforts combining targeted biochemical perturbations and quantitative modeling to specifically address the diverse roles that phase separation processes may play in biology.
Richard She and Daniel F. Jarosz.
Cell. 2018
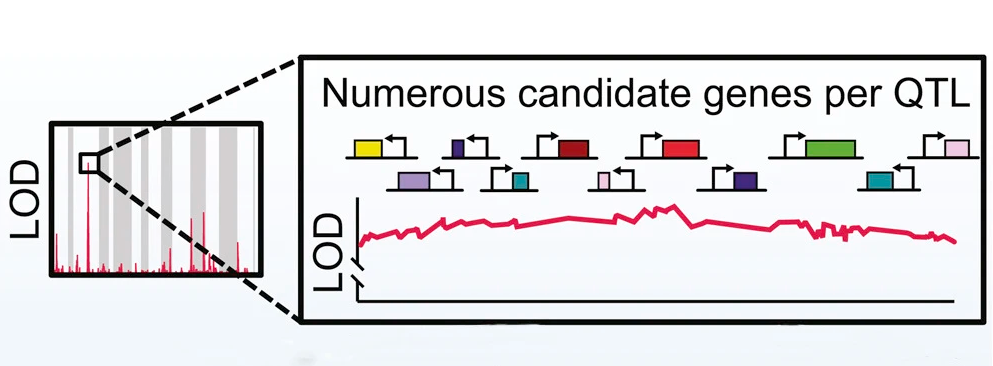
Understanding the sequence determinants that give rise to diversity among individuals and species is the central challenge of genetics. However, despite ever greater numbers of sequenced genomes, most genome-wide association studies cannot distinguish causal variants from linked passenger mutations spanning many genes. We report that this inherent challenge can be overcome in model organisms. By pushing the advantages of inbred crossing to its practical limit in Saccharomyces cerevisiae, we improved the statistical resolution of linkage analysis to single nucleotides. This “super-resolution” approach allowed us to map 370 causal variants across 26 quantitative traits. Missense, synonymous, and cis-regulatory mutations collectively gave rise to phenotypic diversity, providing mechanistic insight into the basis of evolutionary divergence. Our data also systematically unmasked complex genetic architectures, revealing that multiple closely linked driver mutations frequently act on the same quantitative trait. Single-nucleotide mapping thus complements traditional deletion and overexpression screening paradigms and opens new frontiers in quantitative genetics.
Featured in Cell Preview: Meiotic Recombination: Genetics’ Good Old Scalpel. Cell. 2018
Zachary H. Harvey*, Yiwen Chen*, and Daniel F. Jarosz.
Molecular Cell. 2018
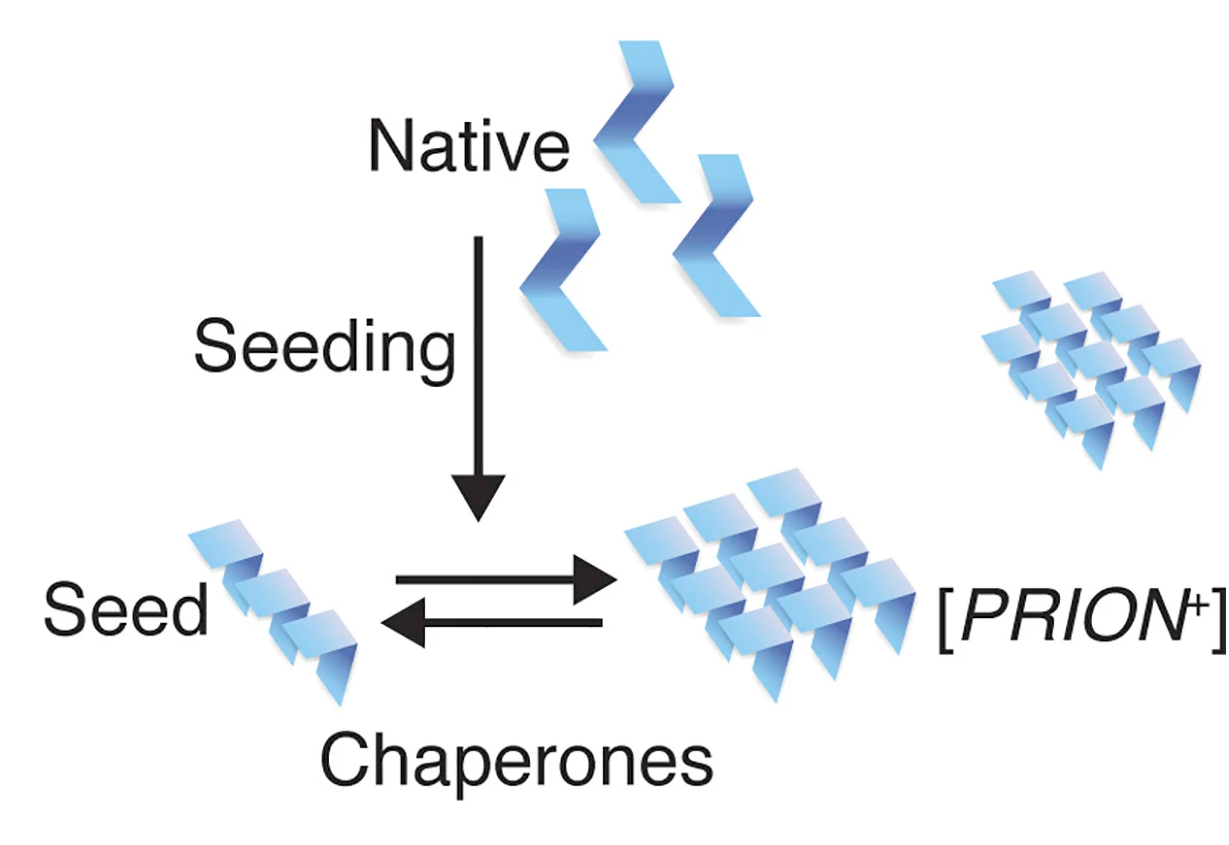
Epigenetics refers to changes in phenotype that are not rooted in DNA sequence. This phenomenon has largely been studied in the context of chromatin modification. Yet many epigenetic traits are instead linked to self-perpetuating changes in the individual or collective activity of proteins. Most such proteins are prions (e.g., [PSI+], [URE3+], [SWI+], [MOT3+], [MPH1+], [LSB+], and [GAR+]), which have the capacity to adopt at least one conformation that self-templates over long biological timescales. This allows them to serve as protein-based epigenetic elements that are readily broadcast through mitosis and meiosis. In some circumstances, self-templating can fuel disease, but it also permits access to multiple activity states from the same polypeptide and transmission of that information across generations. Ensuing phenotypic changes allow genetically identical cells to express diverse and frequently adaptive phenotypes. Although long thought to be rare, protein-based epigenetic inheritance has now been uncovered in all domains of life.
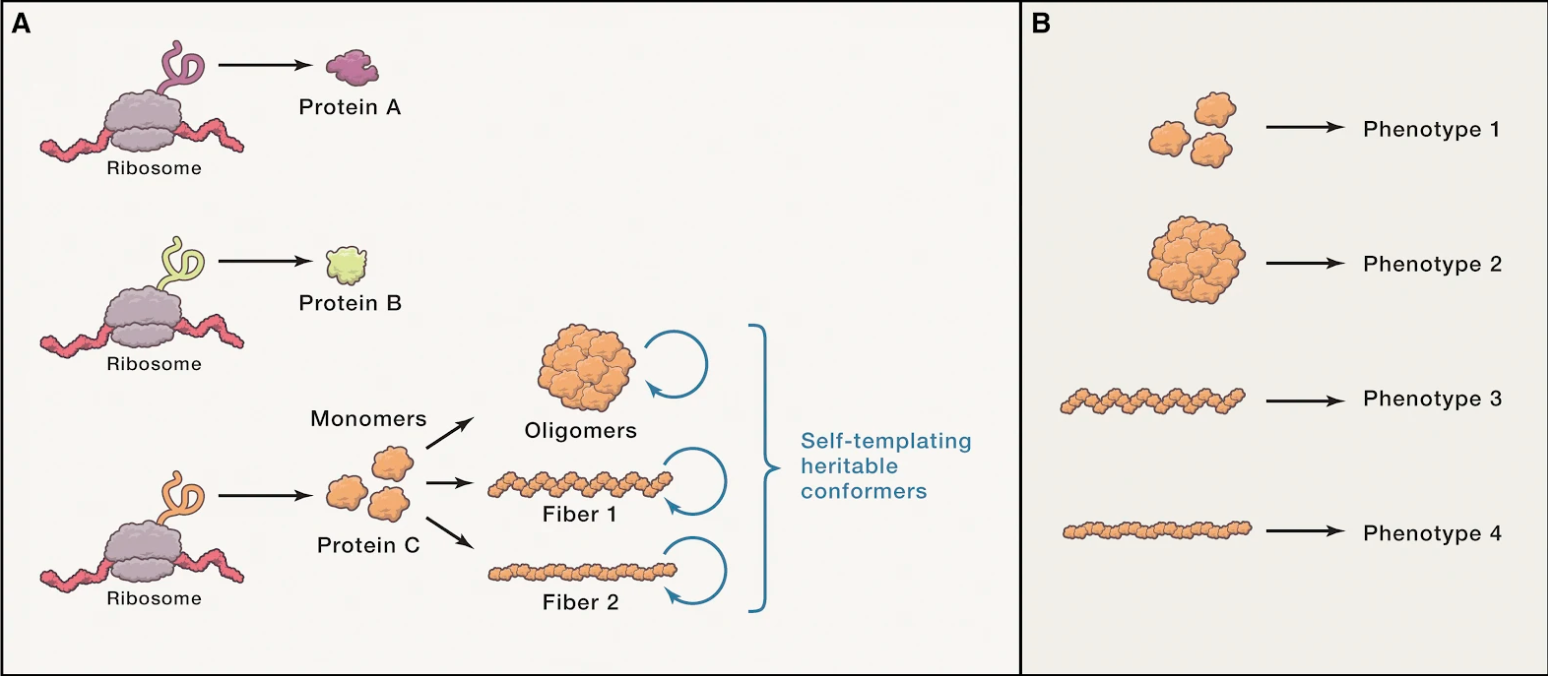
Daniel F. Jarosz and Khurana V.
Cell. 2017
Protein conformational states-from intrinsically disordered ensembles to amyloids that underlie the self-templating, infectious properties of prion-like proteins-have attracted much attention. Here, we highlight the diversity, including differences in biophysical properties, that drive distinct biological functions and pathologies among self-templating proteins. Advances in chemical genomics, gene editing, and model systems now permit deconstruction of the complex interplay between these protein states and the host factors that react to them. These methods reveal that conformational switches modulate normal and abnormal information transfer and that intimate relationships exist between the intrinsic function of proteins and the deleterious consequences of their misfolding.
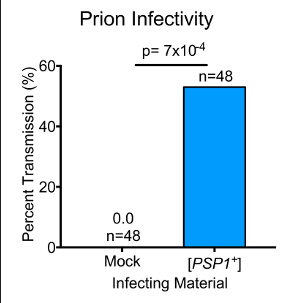
James S. Byers III and Daniel F. Jarosz.
Journal of Visualized Experiments. 2017
The encoding of biological information that is accessible to future generations is generally achieved via changes to the DNA sequence. Long-lived inheritance encoded in protein conformation (rather than sequence) has long been viewed as paradigm-shifting but rare. The best characterized examples of such epigenetic elements are prions, which possess a self-assembling behavior that can drive the heritable manifestation of new phenotypes. Many archetypal prions display a striking N/Q-rich sequence bias and assemble into an amyloid fold. These unusual features have informed most screening efforts to identify new prion proteins. However, at least three known prions (including the founding prion, PrPSc) do not harbor these biochemical characteristics. We therefore developed an alternative method to probe the scope of protein-based inheritance based on a property of mass action: the transient overexpression of prion proteins increases the frequency at which they acquire a self-templating conformation. This paper describes a method for analyzing the capacity of the yeast ORFeome to elicit protein based inheritance. Using this strategy, we previously found that >1% of yeast proteins could fuel the emergence of biological traits that were long-lived, stable, and arose more frequently than genetic mutation. This approach can be employed in high throughput across entire ORFeomes or as a targeted screening paradigm for specific genetic networks or environmental stimuli. Just as forward genetic screens define numerous developmental and signaling pathways, these techniques provide a methodology to investigate the influence of protein-based inheritance in biological processes.
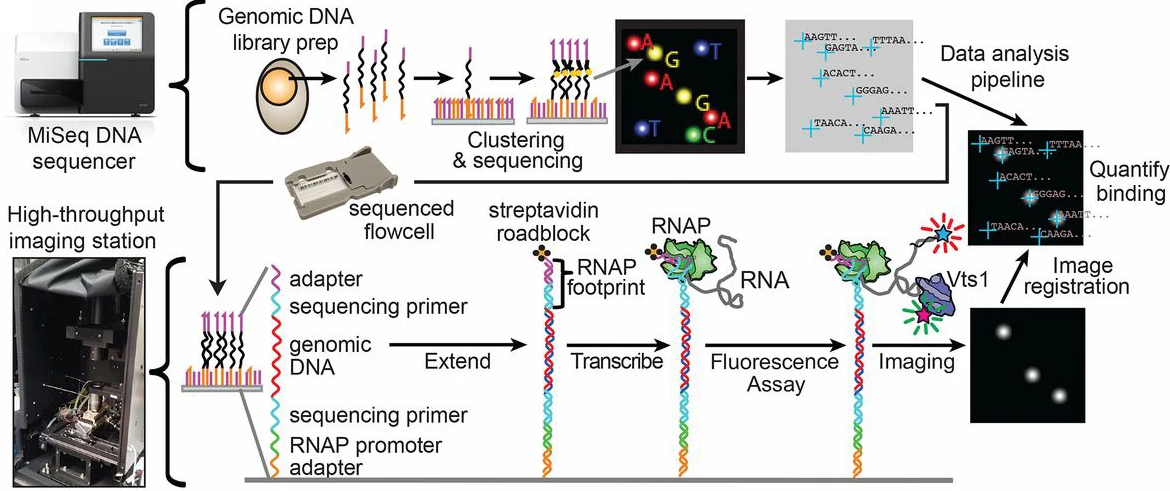
Richard She*, Anupam K. Chakravarty*, Curtis J. Layton*, Lauren M. Chircus, Johan O. Andreasson, Nandita Damaraju, Peter L. McMahon, Jason D. Buenrostro, Daniel F. Jarosz, and William J. Greenleaf.
PNAS. 2017.
RNA-binding proteins (RBPs) control the fate of nearly every transcript in a cell. However, no existing approach for studying these posttranscriptional gene regulators combines transcriptome-wide throughput and biophysical precision. Here, we describe an assay that accomplishes this. Using commonly available hardware, we built a customizable, open-source platform that leverages the inherent throughput of Illumina technology for direct biophysical measurements. We used the platform to quantitatively measure the binding affinity of the prototypical RBP Vts1 for every transcript in the Saccharomyces cerevisiae genome. The scale and precision of these measurements revealed many previously unknown features of this well-studied RBP. Our transcribed genome array (TGA) assayed both rare and abundant transcripts with equivalent proficiency, revealing hundreds of low-abundance targets missed by previous approaches. These targets regulated diverse biological processes including nutrient sensing and the DNA damage response, and implicated Vts1 in de novo gene “birth.” TGA provided single-nucleotide resolution for each binding site and delineated a highly specific sequence and structure motif for Vts1 binding. Changes in transcript levels in vts1Δ cells established the regulatory function of these binding sites. The impact of Vts1 on transcript abundance was largely independent of where it bound within an mRNA, challenging prevailing assumptions about how this RBP drives RNA degradation. TGA thus enables a quantitative description of the relationship between variant RNA structures, affinity, and in vivo phenotype on a transcriptome-wide scale. We anticipate that TGA will provide similarly comprehensive and quantitative insights into the function of virtually any RBP.
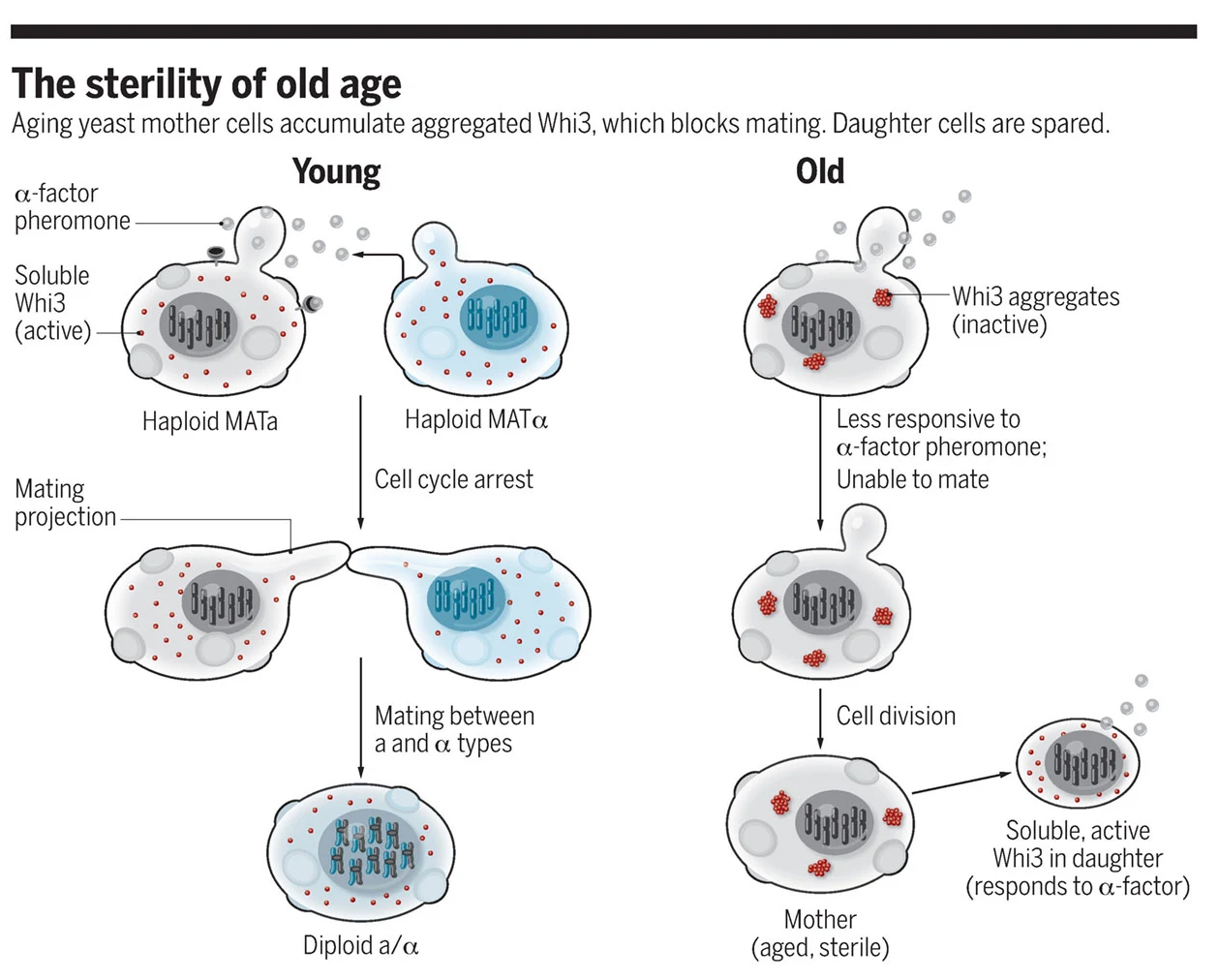
Aaron D. Gitler and Daniel F. Jarosz.
Science. 2017
Is aging ineluctable, or are there genetic programs of aging that could be manipulated to extend life span? Research over the past two decades has provided powerful evidence that aging is indeed regulated by genes that control highly conserved pathways. For example, mutations in single genes in model organisms like flies and worms not only allow these animals to live longer, but rejuvenate them as well. Even the single-celled budding yeast Saccharomyces cerevisiae ages, and specific genes likewise control this process. Indeed, one of the most powerful genetic manipulations to extend life span was discovered in yeast. The histone deacetylase silent information regulator 2 (Sir2) is required for repressing the transcription of certain mating-type loci, telomeres, and ribosomal DNA. The latter had been linked to aging in yeast, inspiring studies that revealed Sir2’s importance for this process. Subsequent work in yeast and animal models established that changes in Sir2 activity are responsible for much of the life span–extending effects of caloric restriction. On page 1184 of this issue, Schlissel et al. report that a particular facet of aging, which had long been attributed to age-dependent changes in Sir2 function, is caused by a new mechanism.
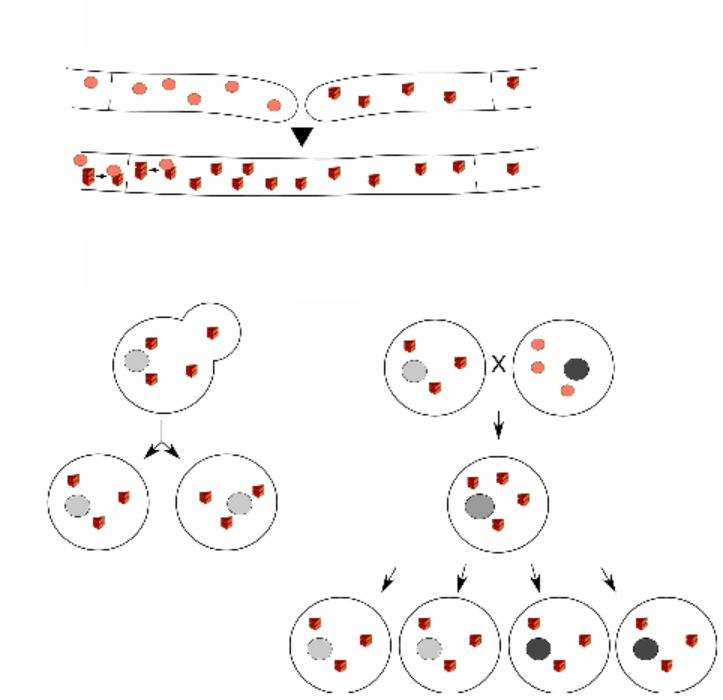
Sven J. Saupe, Daniel F. Jarosz, and Heather L. True.
Microbiology Spectrum. 2016
Prions are infectious protein polymers that have been found to cause fatal diseases in mammals. Prions have also been identified in fungi (yeast and filamentous fungi), where they behave as cytoplasmic non-Mendelian genetic elements. Fungal prions correspond in most cases to fibrillary β-sheet-rich protein aggregates termed amyloids. Fungal prion models and, in particular, yeast prions were instrumental in the description of fundamental aspects of prion structure and propagation. These models established the “protein-only” nature of prions, the physical basis of strain variation, and the role of a variety of chaperones in prion propagation and amyloid aggregate handling. Yeast and fungal prions do not necessarily correspond to harmful entities but can have adaptive roles in these organisms.
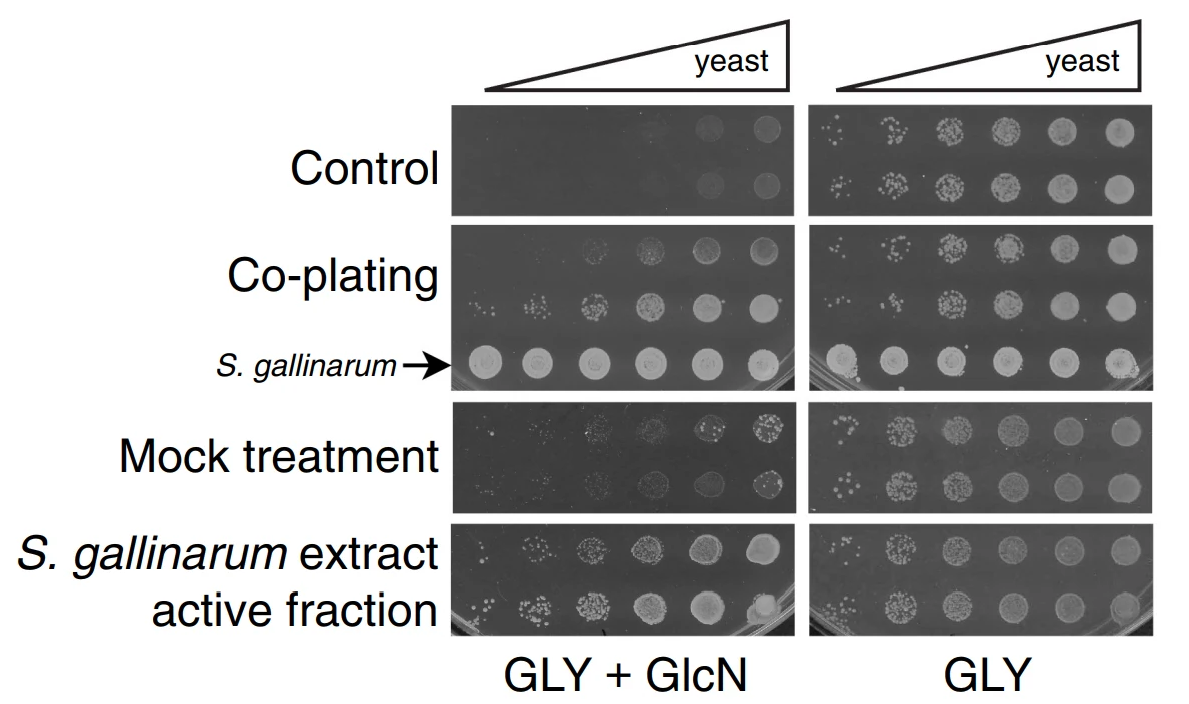
David M. Garcia, David Dietrich, Jon Clardy, and Daniel F. Jarosz.
eLife. 2016
Robust preference for fermentative glucose metabolism has motivated domestication of the budding yeast Saccharomyces cerevisiae. This program can be circumvented by a protein-based genetic element, the [GAR+] prion, permitting simultaneous metabolism of glucose and other carbon sources. Diverse bacteria can elicit yeast cells to acquire [GAR+], although the molecular details of this interaction remain unknown. Here we identify the common bacterial metabolite lactic acid as a strong [GAR+] inducer. Transient exposure to lactic acid caused yeast cells to heritably circumvent glucose repression. This trait had the defining genetic properties of [GAR+], and did not require utilization of lactic acid as a carbon source. Lactic acid also induced [GAR+]-like epigenetic states in fungi that diverged from S. cerevisiae ~200 million years ago, and in which glucose repression evolved independently. To our knowledge, this is the first study to uncover a bacterial metabolite with the capacity to potently induce a prion.
Featured in:
An Acid Tale of Prion Formation. eLIFE. 2016
Editor’s Choice: Bacteria Stop Fermentation with Lactic Acid. Science Signaling. 2016
How prions help yeast feast on a wide range of sugars. Stanford Scope Blog. 2016
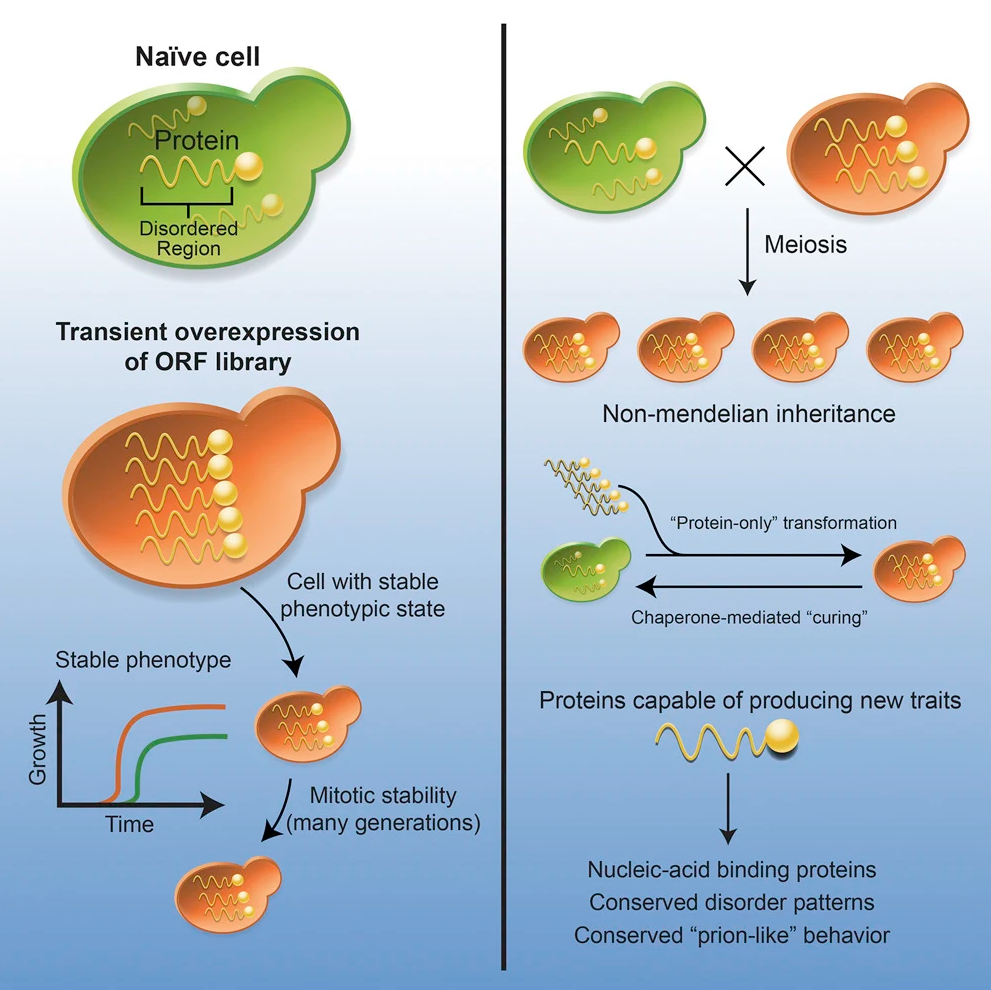
Sohini Chakrabortee*, James S. Byers III*, Sandra Jones, David M. Garcia, Bhupinder Bhullar, Amelia Chang, Richard She, Laura Lee, Brayon Fremin, Susan Lindquist, and Daniel F. Jarosz.
Cell. 2016
Prions are a paradigm-shifting mechanism of inheritance in which phenotypes are encoded by self-templating protein conformations rather than nucleic acids. Here, we examine the breadth of protein-based inheritance across the yeast proteome by assessing the ability of nearly every open reading frame (ORF; ∼5,300 ORFs) to induce heritable traits. Transient overexpression of nearly 50 proteins created traits that remained heritable long after their expression returned to normal. These traits were beneficial, had prion-like patterns of inheritance, were common in wild yeasts, and could be transmitted to naive cells with protein alone. Most inducing proteins were not known prions and did not form amyloid. Instead, they are highly enriched in nucleic acid binding proteins with large intrinsically disordered domains that have been widely conserved across evolution. Thus, our data establish a common type of protein-based inheritance through which intrinsically disordered proteins can drive the emergence of new traits and adaptive opportunities.
Featured in:
Remembering the Past: A New Form of Protein-Based Inheritance. Cell. 2016
Prions can pass on beneficial traits, study finds. Stanford Medicine News Center. 2016
Revising the meaning of ‘prion.’ Science Daily. 2016
David M. Garcia and Daniel F. Jarosz.
FEMS Yeast Research. 2014
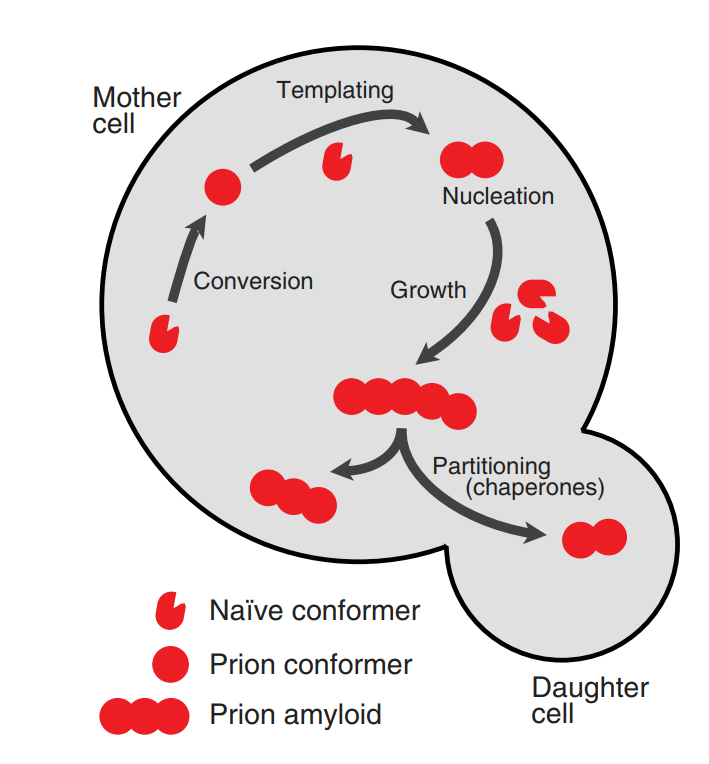
Prions are proteins that convert between structurally and functionally distinct states, at least one of which is self-perpetuating. The prion fold templates the conversion of native protein, altering its structure and function, and thus serves as a protein-based element of inheritance. Molecular chaperones ensure that these prion aggregates are divided and faithfully passed from mother cells to their daughters. Prions were originally identified as the cause of several rare neurodegenerative diseases in mammals, but the last decade has brought great progress in understanding their broad importance in biology and evolution. Most prion proteins regulate information flow in signaling networks, or otherwise affect gene expression. Consequently, switching into and out of prion states creates diverse new traits – heritable changes based on protein structure rather than nucleic acid. Despite intense study of the molecular mechanisms of this paradigm-shifting, epigenetic mode of inheritance, many key questions remain. Recent studies in yeast that support the view that prions are common, often beneficial elements of inheritance that link environmental stress to the appearance of new traits.
Daniel F. Jarosz*, Jessica C.S. Brown*, Gordon A. Walker, Manoshi S. Datta, W. Lloyd Ung, Alex K. Lancaster, Assaf Rotem, Amelia Chang, Gregory A. Newby, David A. Weitz, Linda F. Bisson, and Susan Lindquist.
Cell. 2014
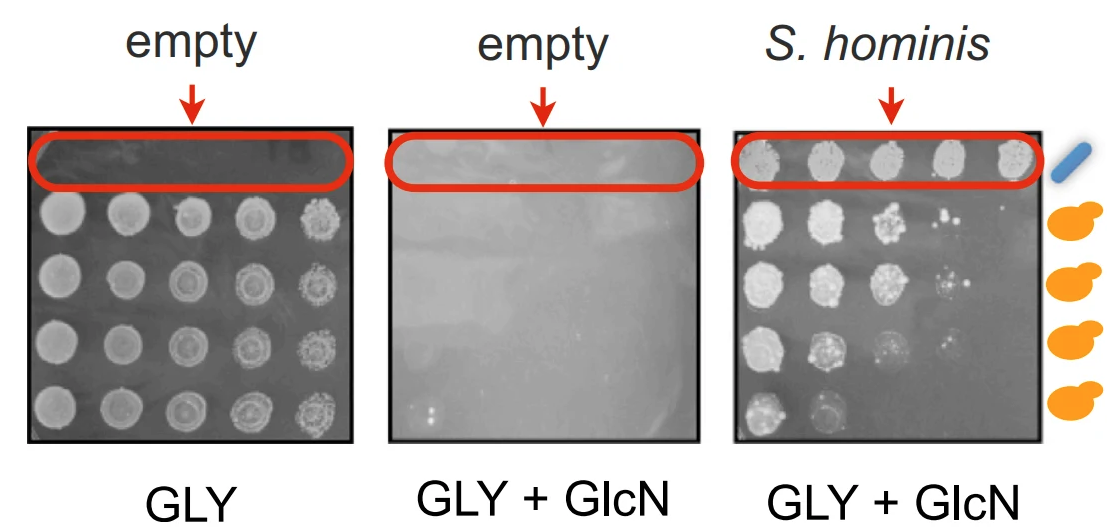
In experimental science, organisms are usually studied in isolation, but in the wild, they compete and cooperate in complex communities. We report a system for cross-kingdom communication by which bacteria heritably transform yeast metabolism. An ancient biological circuit blocks yeast from using other carbon sources in the presence of glucose. [GAR(+)], a protein-based epigenetic element, allows yeast to circumvent this “glucose repression” and use multiple carbon sources in the presence of glucose. Some bacteria secrete a chemical factor that induces [GAR(+)]. [GAR(+)] is advantageous to bacteria because yeast cells make less ethanol and is advantageous to yeast because their growth and long-term viability is improved in complex carbon sources. This cross-kingdom communication is broadly conserved, providing a compelling argument for its adaptive value. By heritably transforming growth and survival strategies in response to the selective pressures of life in a biological community, [GAR(+)] presents a unique example of Lamarckian inheritance.
Daniel F. Jarosz*, Alex K. Lancaster*, Jessica C.S. Brown, and Susan Lindquist.
Cell. 2014
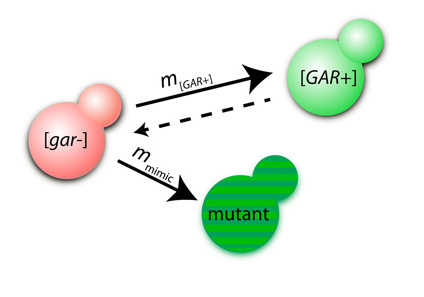
[GAR+] is a protein-based element of inheritance that allows yeast (Saccharomyces cerevisiae) to circumvent a hallmark of their biology: extreme metabolic specialization for glucose fermentation. When glucose is present, yeast will not use other carbon sources. [GAR+] allows cells to circumvent this “glucose repression.” [GAR+] is induced in yeast by a factor secreted by bacteria inhabiting their environment. We report that de novo rates of [GAR+] appearance correlate with the yeast’s ecological niche. Evolutionarily distant fungi possess similar epigenetic elements that are also induced by bacteria. As expected for a mechanism whose adaptive value originates from the selective pressures of life in biological communities, the ability of bacteria to induce [GAR+] and the ability of yeast to respond to bacterial signals have been extinguished repeatedly during the extended monoculture of domestication. Thus, [GAR+] is a broadly conserved adaptive strategy that links environmental and social cues to heritable changes in metabolism.
Featured in Monoculture Breeds Poor Social Skills. Cell, 2014
James S. Byers III and Daniel F. Jarosz.
PLOS Pathogens. 2014
Prions are “infectious” misfolded protein states that can template their self-perpetuating conformations onto other molecules of the same type. This unusual folding landscape drives a paradigm-shifting mechanism of inheritance based on changes in protein conformation rather than changes in nucleic acid. The first prion discovered, Prion Protein (PrP), is the causal agent of several human neurodegenerative diseases, including kuru and Creutzfeldt-Jakob. This history has engendered the widespread perception that prions are inherently pathogenic. However, many additional prions have now been found in other eukaryotes in which they influence diverse biological processes and can produce beneficial traits. The most well-characterized of these are found in Saccharomyces cerevisiae and other fungi, such as Podospora anserina.

Nicolas Rohner*, Daniel F. Jarosz, Johanna E. Kowalko, Masato Yoshizawa, William R. Jeffery, Richard L. Borowsky, Susan Lindquist, and Clifford J. Tabin.
Science. 2013
In the process of morphological evolution, the extent to which cryptic, preexisting variation provides a substrate for natural selection has been controversial. We provide evidence that heat shock protein 90 (HSP90) phenotypically masks standing eye-size variation in surface populations of the cavefish Astyanax mexicanus. This variation is exposed by HSP90 inhibition and can be selected for, ultimately yielding a reduced-eye phenotype even in the presence of full HSP90 activity. Raising surface fish under conditions found in caves taxes the HSP90 system, unmasking the same phenotypic variation as does direct inhibition of HSP90. These results suggest that cryptic variation played a role in the evolution of eye loss in cavefish and provide the first evidence for HSP90 as a capacitor for morphological evolution in a natural setting.
Featured in:
An eye for cryptic variation. Nature Reviews Genetics. 2013
How a Fish Unleashed Its Evolutionary Potential and Went Blind. National Geographic Science Blog. 2013
Randal Halfmann*, Daniel F. Jarosz*, Sandra K. Jones, Amelia Chang, Alex K. Lancaster, and Susan Lindquist.
Nature. 2012
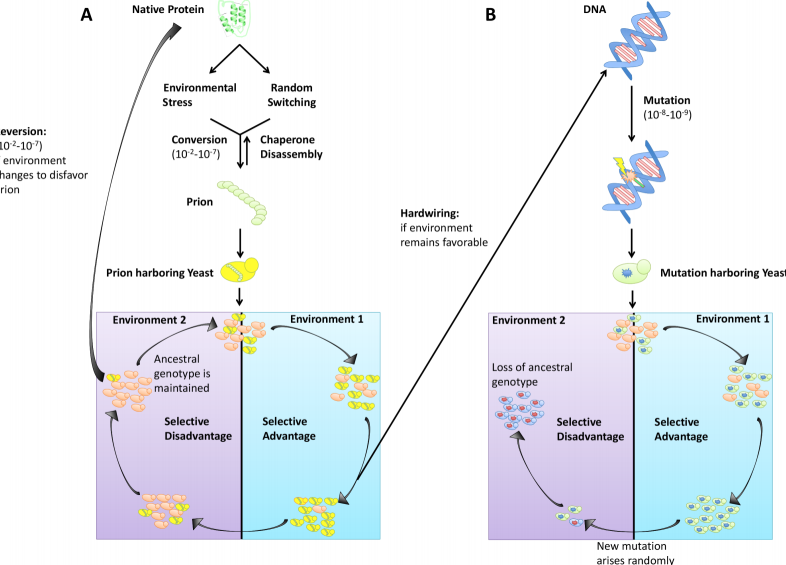
The self-templating conformations of yeast prion proteins act as epigenetic elements of inheritance. Yeast prions might provide a mechanism for generating heritable phenotypic diversity that promotes survival in fluctuating environments and the evolution of new traits. However, this hypothesis is highly controversial. Prions that create new traits have not been found in wild strains, leading to the perception that they are rare ‘diseases’ of laboratory cultivation. Here we biochemically test approximately 700 wild strains of Saccharomyces for [PSI+] or [MOT3+], and find these prions in many. They conferred diverse phenotypes that were frequently beneficial under selective conditions. Simple meiotic re-assortment of the variation harboured within a strain readily fixed one such trait, making it robust and prion-independent. Finally, we genetically screened for unknown prion elements. Fully one-third of wild strains harboured them. These, too, created diverse, often beneficial phenotypes. Thus, prions broadly govern heritable traits in nature, in a manner that could profoundly expand adaptive opportunities.
Featured in:
Research Highlights. Nature Microbiology. 2012
Yeast find use for misfolded proteins. Science News. 2012
Propitious prions. The Scientist. 2012
Prions point to a new style of evolution. New Scientist. 2012
Prion: pour le pire… et le meilleur. Science & Vie. 2012
Daniel F. Jarosz, and Susan Lindquist.
Science. 2010
Daniel F. Jarosz*, Mikko Taipale*, and Susan Lindquist
Annual Review of Genetics 44, 189-216 (2010) *Equal authorship
Mikko Taipale*, Daniel F. Jarosz*, and Susan Lindquist
Nature Reviews Molecular Cell Biology 11, 515-528 (2010) *Equal authorship
Daniel F. Jarosz, Susan E. Cohen, James C. Delaney, John M. Essigmann, and Graham C. Walker
PNAS 106, 21137-21142 (2010)
Veronica G. Godoy*, Daniel F. Jarosz*, Sharotka Simon, Alexei Abyzov, Valentin Ilyin, and Graham C. Walker
Molecular Cell 28,1058-1070 (2007) *Equal authorship
Daniel F. Jarosz, Veronica G. Godoy, and Graham C. Walker
Cell Cycle 6, 818-822 (2007)
William L. Neeley, Sarah Delaney, Yuriy O. Alekseyev, Daniel F. Jarosz
Journal of Biological Chemistry 282, 12741-12748 (2007)
Daniel F. Jarosz, Penny J. Beuning, Susan E. Cohen, and Graham C. Walker
Trends in Microbiology 15, 70-77 (2007)
Penny J. Beuning, Sharotka M. Simon, Veronica G. Godoy, Daniel F. Jarosz, and Graham C. Walker
Methods in Enzymology 408, 318-339 (2006)
Veronica G. Godoy, Daniel F. Jarosz, Fabianne L. Walker, Lyle A. Simmons, and Graham C. Walker
EMBO Journal 25, 868-879 (2006)
Daniel F. Jarosz*, Veronica G. Godoy*, James C. Delaney, John M. Essigmann, and Graham C. Walker
Nature, 439, 225-228 (2006) *Equal authorship
Daniel F. Jarosz, Jessica C.S. Brown, and Susan Lindquist.
August 1, 2012.
Information regarding databases will be updated shortly in this section.
