Some biological systems can remain unaltered for long periods, whereas others that are genetically identical undergo rapid diversification. This paradox lies at the heart of how neurons can be killed by improper expression of a single aggregation-prone protein, how cancer cells can tolerate accumulating mutation burden, and how disease-associated mutations have devastating consequences in some individuals, but no effect in others. We seek to understand mechanisms driving this behavior, and to do so we employ multidisciplinary approaches ranging from chemical biology to systems-level quantitative genetics and use models as diverse as baker’s yeast and the African turquoise killifish.
Understanding the origins of diversity among individuals and species is a central challenge of genetics. Yet most genome-wide association studies cannot distinguish causal variants from linked passenger mutations. We have combined theory and experiment to overcome this challenge (She and Jarosz, Cell, 2018), pushing inbred crossing to its practical limit in Saccharomyces cerevisiae to improve the resolution of linkage analysis from kilobases to single nucleotides. This ‘super-resolution’ approach has led to the surprising finding that missense, synonymous, and cis-regulatory variants collectively give rise to phenotypic diversity, with comparable effect sizes. Most traits are extremely complex – driven by multiple coding and non-coding variants alike – and closely linked driver mutations frequently act on the same trait. This approach complements traditional genetic screening paradigms, opening new frontiers in quantitative genetics and providing valuable lessons that can be extended to other organisms.
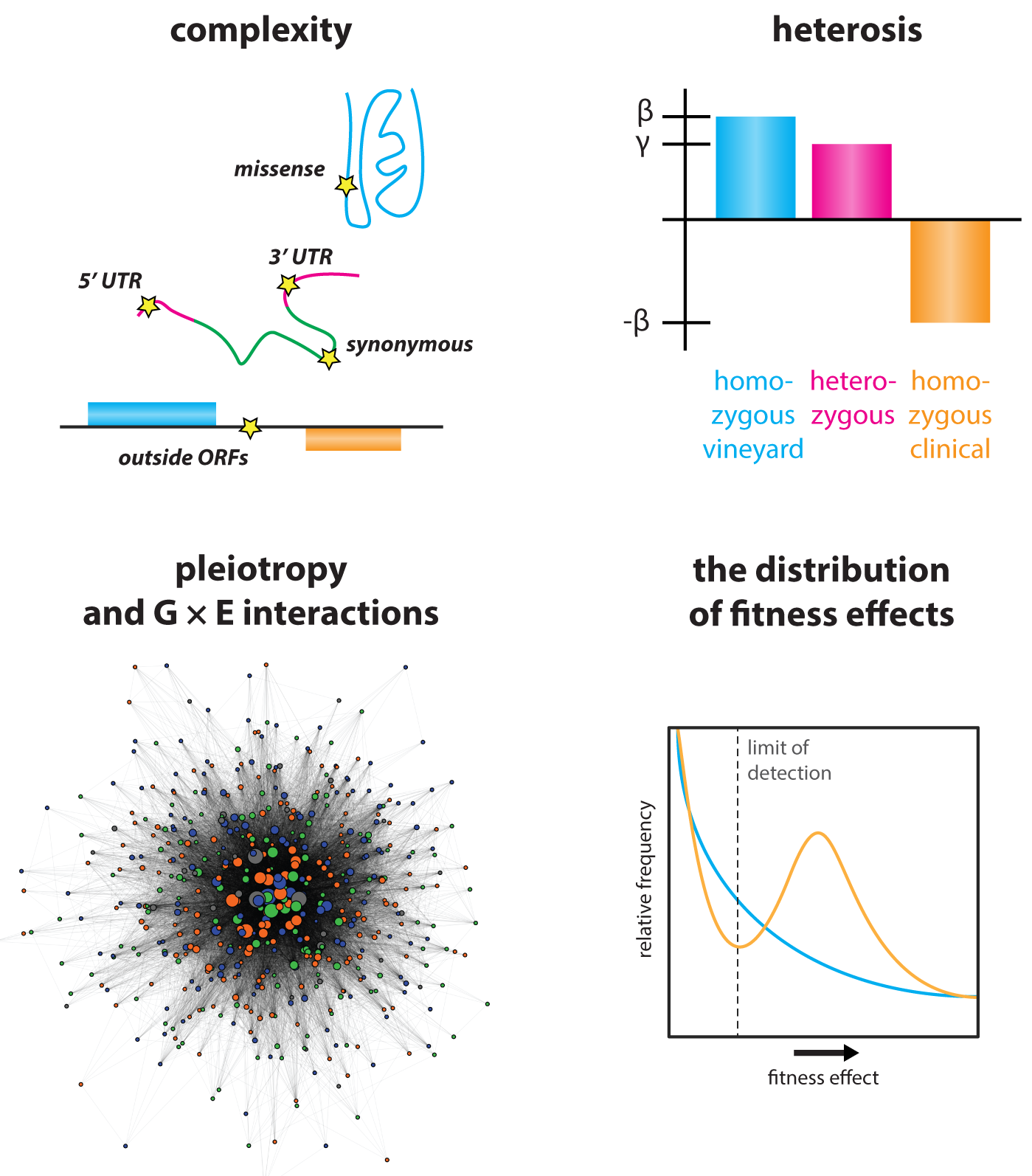
The importance of synonymous variants – Our discovery that natural synonymous variants frequently produce large phenotypic effects challenges prevailing wisdom. Most measures of selection and pragmatic assumptions in clinical genetics assume that synonymous mutations are neutral. Exploring possible mechanisms, we found that synonymous mutations with large phenotypic impact exerted strong effects on codon adaptation index early in ORFs, suggesting a possible role in regulating translation. Consistent with this inference, causal synonymous mutations often had a strong influence on protein levels. Missense mutations, by contrast, affect codon adaptation index more modestly, and simulations revealed this property to be a general feature of codon usage in organisms across the tree of life, despite extreme variation in codon usage frequency. Our findings suggest these variants work orthogonally: missense mutations act to diversify protein function, whereas synonymous mutations can tune expression levels, acting as a form of regulatory variation within ORFs.
Pervasive selection on standing genetic variation – Quantitative genetics often aims to understand how organisms evolved. Yet it remains unclear whether the variants identified are exemplary of evolution. Our finding that synonymous variants frequently have large phenotypic impact motivated us to develop an alternative metric for selection, based on repeated emergence in phylogenies. Remarkably, and contrary to drift-centric theories, we have found that many natural genetic variants exhibit patterns inconsistent with, likely attributable to both homoplasy and balancing selection on ancestral polymorphism. Variants that emerged repeatedly were more likely to have done so in genetic backgrounds isolated from the same ecological niche, further underscoring the power of super-resolution mapping in understanding evolutionary adaptation.
New technologies – Although sequencing approaches have revolutionized our understanding of gene control, few techniques combine transcriptome-wide throughput and biophysical precision. In collaboration with Will Greenleaf (Stanford Genetics) we have generated a uniform, highly redundant transcribed genome array (TGA) on an Illumina sequencing chip in an effort to understand the function of RNA binding proteins on a transcriptome-wide scale. We harnessed this technology to identify hundreds of new targets of the evolutionary ancient RNA binding protein Smaug/Vts1 with nucleotide resolution, and direct measurements of affinity and dissociation. Our data supplant prior knowledge of substrate recognition, which was thought to occur via 3’-UTR binding. We found that destabilization could occur from binding throughout the ORF. This technology also revealed unknown roles for Vts1 in the induction of stress responses and the birth of new genes and is now providing similarly comprehensive and quantitative insight into many other RNA binding proteins.
↑ Back To Top
Most biological molecules are synthesized as long linear polymers that must adopt complex three-dimensional structures to function. This folding process can be challenging in the crowded intracellular milieu, particularly for mutant proteins. Molecular chaperones, which help these polymers to fold, can thus have a critical influence on evolution and disease. Much of our work in this area has focused on Hsp90, an essential ATP-driven chaperone that assists the folding of proteins that regulate growth and development. In humans, these ‘client’ kinases and transcription factors play critical roles in signaling and cancer. Using a combination of quantitative genetics and chemical biology, our work has established that Hsp90 fundamentally re-shapes the map between genotype and phenotype. Most remarkably, we uncovered traces of Hsp90’s effects in the correlation between genotype and phenotype in sequenced genomes, pointing to importance of this mechanism in shaping evolution. Together our studies have transformed an intriguing hypothesis into a very likely mechanism for environmental regulation of evolutionary change in both normal biology and disease states. Recently, we have extended this line of research to uncover a stress response that hypermutating eukaryotic cells exploit to evolve and proliferate; targeting pathways engaged in this response selectively kills highly mutating cells.
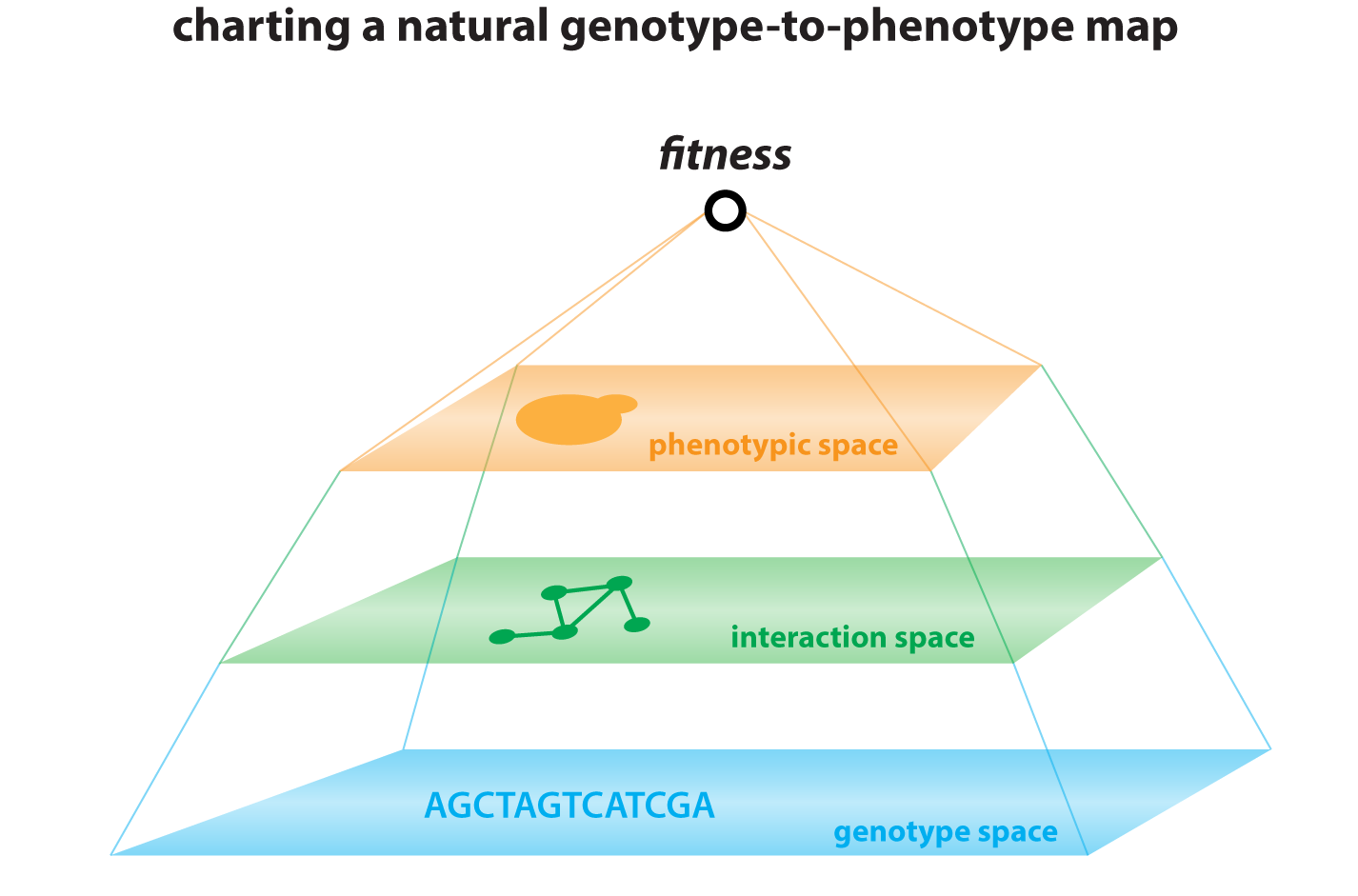
Molecular origins of genetic complexity – The intrinsic complexity of the genotype-to-phenotype relationship has been appreciated since Mendel’s work was rediscovered at the beginning of the 20th century. But there remains a fundamental gap between evident statistical patterns of heredity and an understanding of their molecular origins. We have begun to break this logjam, generating mapping populations of >18,000 sequenced homozygous and heterozygous diploids, providing low linkage disequilibrium and, for the first time, a greater number of genotyped individuals than segregating polymorphisms. This allowed us to map all linear contributors to phenotype, providing answers to many longstanding questions in heredity. Our data have revealed that natural genetic networks are topologically distinct from those defined by precise deletions, and that both pleiotropic and gene x environment interactions abound in them. Surprisingly, the effect sizes of pleiotropic variants are correlated in different environments, even when their effects were opposing. This suggests that such alleles represent central regulatory nodes of the genotype-to-phenotype map. Indeed, highly pleiotropic variants affected key signaling hubs and, intriguingly, often altered intrinsically disordered tracts within these proteins. Such loci are highly polymorphic in nature, implicating them in rapid phenotypic diversification.
Macromolecular folding links phenotypes to environmental change – Hsp90 is an essential molecular chaperone that catalyzes the folding of client protein kinases, transcription factors, and ubiquitin ligases with critical functions in growth and development. Like other chaperones, its activity is regulated by environmental stress. My postdoctoral work established that Hsp90 has a broad effect on the genotype-to-phenotype map and has likely influenced evolutionary processes in fungi. A collaboration with Cliff Tabin (Harvard Genetics) suggested that this mechanism has also coupled environmental change to the rapid emergence of adaptive traits in Mexican cavefish. Yet the genetic variants involved remained enigmatic. Our super-resolution mapping technology have allowed us to overcome this barrier, identifying scores of natural variants impacted by chaperone function. These are often in portions of client proteins thought to interact with Hsp90. But others arose in non-coding regulatory sequences, pointing to previously unappreciated mechanisms through which protein chaperones can influence the acquisition of new traits.
We have also investigated how the folding of another biological polymer – RNA –influences phenotypic diversification. Like proteins, RNAs often must acquire complex structures to exert their biological functions. We have found that the phenotypic consequences of many natural genetic variants, and hence their adaptive value, can be strongly influenced by the stress-regulated RNA chaperone Lhp1/LARP. Traits encoded by some variants are potentiated by the chaperone, whereas others are buffered. Both coding and non-coding variants are affected, defining new interface of macromolecular folding, environmental stress, and ensuing biological phenotype.
A stress response that allows highly mutated cells to survive, evolve, and proliferate – Rapid mutation fuels the evolution of many cancers and pathogens. Most ensuing genetic variation is detrimental, but cells can limit its cost. We have investigated this behavior, propagating hypermutating lineages to create independent populations harboring thousands of distinct genetic variants. Mutation rate and spectrum remained unchanged throughout the experiment, yet lesions that arose early were more deleterious than those that arose later. Although the lineages share no mutations in common, each mounted a similar transcriptional response to mutation burden, which we term EMBR (for eukaryotic mutation burden response). The ~200 proteins involved form a highly connected network that has not previously been identified. Inhibiting this response increased the cost of accumulated mutations, selectively killing highly mutated cells. A similar gene expression program, linked to survival, exists in hypermutating human cancers. Our data thus define a conserved stress response that buffers the cost of accumulating genetic lesions and suggest that this network could be targeted therapeutically and potentially harnessed in directed evolution, where protein instability and other pitfalls of mutation impose severe constraints.
↑ Back To Top
Although best known as the causal agents of spongiform encephalopathies, the self-templating protein conformations known as prions can also function as protein-only elements of inheritance that are stable over long biological timescales. However, the adaptive value of this form of epigenetics ‘beyond the chromosome’ had long remained uncertain. We have carried out a suite of biochemical and phenotypic screens that have suggested that these elements of inheritance are common in nature, where they frequently confer beneficial phenotypes ranging from drug resistance to changes in social behavior. More recently we have extended this work to identify fifty additional prion-like molecular memories in eukaryotic proteomes and are identifying their triggers and characterizing their influence on gene expression.
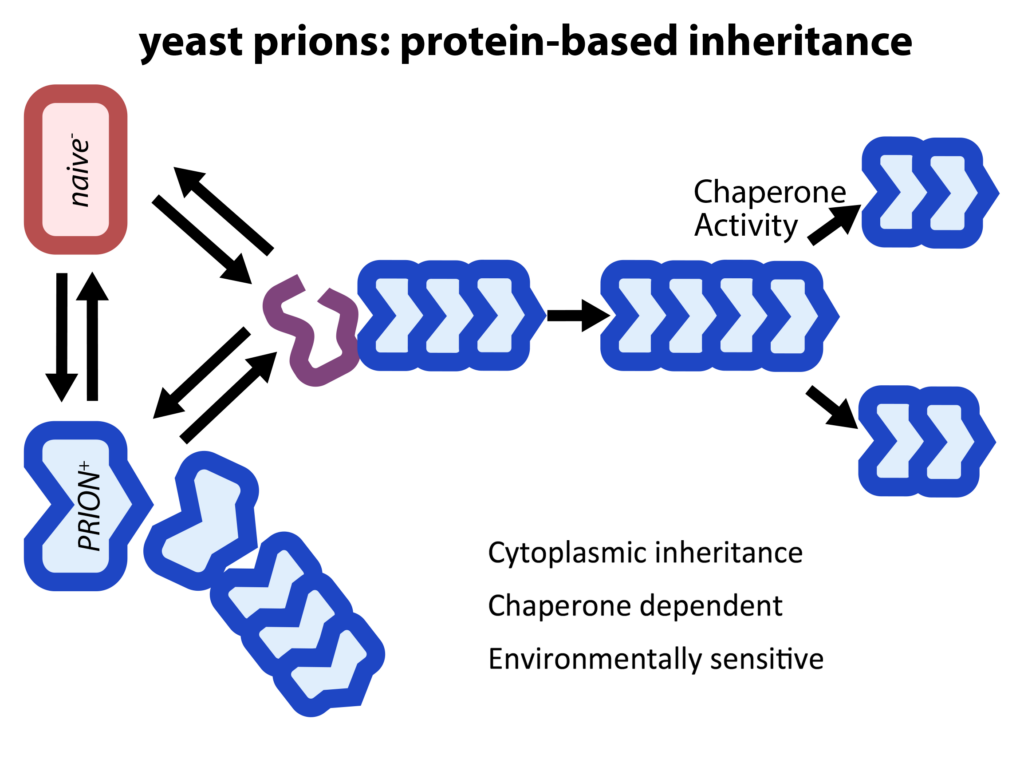
Broadening the prion concept: A new form of protein-based epigenetics – Nearly 30% of eukaryotic proteins are intrinsically disordered: they do not adopt a fixed structure. This dark matter of the proteome is clearly important. Many disease mutations occur within intrinsically disordered regions (IDRs) and most transcription factors (TFs) and RNA binding proteins (RBPs) harbor them. Yet in many studies IDRs have been removed for convenience. IDRs are becoming increasingly well recognized as drivers of phase separation. We have discovered that they can also fuel a form of beneficial, non-amyloid self-assembly that is stable over long biological timescales. Such epigenetics ‘beyond the chromosome’ can be robustly induced by environmental stimuli, and is often hidden in plain sight, controlling fundamental decisions in growth, metabolism, and gene regulation.
Self-assembly into active non-amyloid particles – Our discoveries in this area began with a systematic examination of protein-based inheritance across the S. cerevisiae proteome. Transient overexpression of ~50 proteins created traits that were heritable many generations after expression returned to normal. These traits are generally beneficial and had prion-like inheritance patterns. They were efficiently transmitted to naïve cells with lysates subjected to extensive nuclease digestion, consistent with protein-only inheritance. Strikingly, most proteins we identified are not known prions and did not form amyloid. Instead, they are enriched in nucleic acid binding proteins with large IDRs that have been conserved from yeast to humans, where orthologs often retain the capacity to self-template. Our findings have established a new and common type of protein-based inheritance through which intrinsically disordered proteins can drive the emergence of new traits and adaptive opportunities.
In contrast to archetypal amyloid prions, which often impair a protein’s native function, our in vitro studies have revealed that condensation of these proteins frequently hyperactivates their function. Although they appear round, condensates have physicochemical properties that distinguish them from droplets formed by liquid-liquid phase separation and from fibrillar aggregates. Instead they form gel-like particles that, even when generated in vitro, can transform the phenotype of naïve cells and their descendants. Our findings establish the conserved capacity of IDRs to couple phase separation and protein-based inheritance in a new mechanism of epigenetics: non-amyloid self-assembly that heritably activates protein function.
Protein self-assembly in gene control – Enrichment for nucleic acid binding activity among the proteins we discovered has led us to test whether self-assembly could drive new interpretations of genetic information, complementing conventional epigenetics. We have investigated four representative examples: [MIX+], formed by a DNA helicase; [ESI+], formed by the Snt1 scaffold of the Set3C histone deacetylase complex; [RLM1+], formed by a TF; and [SMAUG+], formed by an RBP. [MIX+] increased meiotic crossovers and improved survival in DNA damaging stresses; several robustly induced [MIX+] acquisition. In genetic crosses [MIX+] increased phenotypic diversity in meiotic progeny, suggesting that it is a quasi-Lamarckian factor that can fuel genetic and phenotypic diversification alike. [ESI+], formed by self-templating conformational conversion of the Set3C histone deacetylase scaffold Snt1, activates silent chromatin by elevating histone acetylation in sub-telomeric regions. This prion, and potentially others formed by chromatin modifiers, thus provide a means for transgenerational inheritance of altered chromatin states. RNA-seq of isogenic [RLM1+] and [rlm1-] cells has revealed an expanded transcriptional program in this prion state. The capacity to self-template was conserved in Rlm1’s human homolog Mef2. [SMAUG+] enhances decay of target RNAs, driven by improved binding and expanded substrate. Metazoan homologs, which coordinate the maternal-to-zygotic transition in development also self-template. Collectively our findings, and enrichment of similar disordered sequences in proteins involved in gene control, illuminate a new tier of regulation in which prion-like conformational conversion can rewire regulatory circuitry.
Mechanisms of induction – Although they are often grown in monoculture in the laboratory, in nature organisms live in complex communities. We discovered a cross-kingdom communication that induces a heritable, prion-based transformation of fungal metabolism. [GAR+] controls whether cells ferment glucose to quickly produce energy, or instead respire to harness its full ATP-generating potential (. Diverse bacteria secrete a factor that elicits [GAR+] in neighboring fungal cells. [GAR+] yeast produce less ethanol, bestowing an advantage to inducing bacteria. [GAR+] cells proliferate more rapidly on complex carbohydrates, providing an advantage in long-term co-culture with bacteria. The bacterial trigger is high concentrations of lactic acid, offering a potential molecular explanation for Louis Pasteur’s classic findings linking lactic acid bacteria to failed fermentations. This is the first known robust molecular prion inducer, and the mutualism is broadly conserved, providing a compelling argument for its adaptive value.
The robust induction of [GAR+] led us to investigate whether other prions might also be regulated. We noted that IDRs in Snt1 important for [ESI+] induction harbor multiple phosphorylation sites controlled by Cdk1. Remarkably, phosphomimic mutations (and Cdk1 induction via nocodazole arrest) induced [ESI+] in nearly all cells, and the prion remained stable even after the original phosphorylation decayed. Our findings underscore how conformational replication inherent to protein self-assembly can amplify transient signaling events, establishing stable ‘memories’ of past stimuli that persist over long biological timescales.
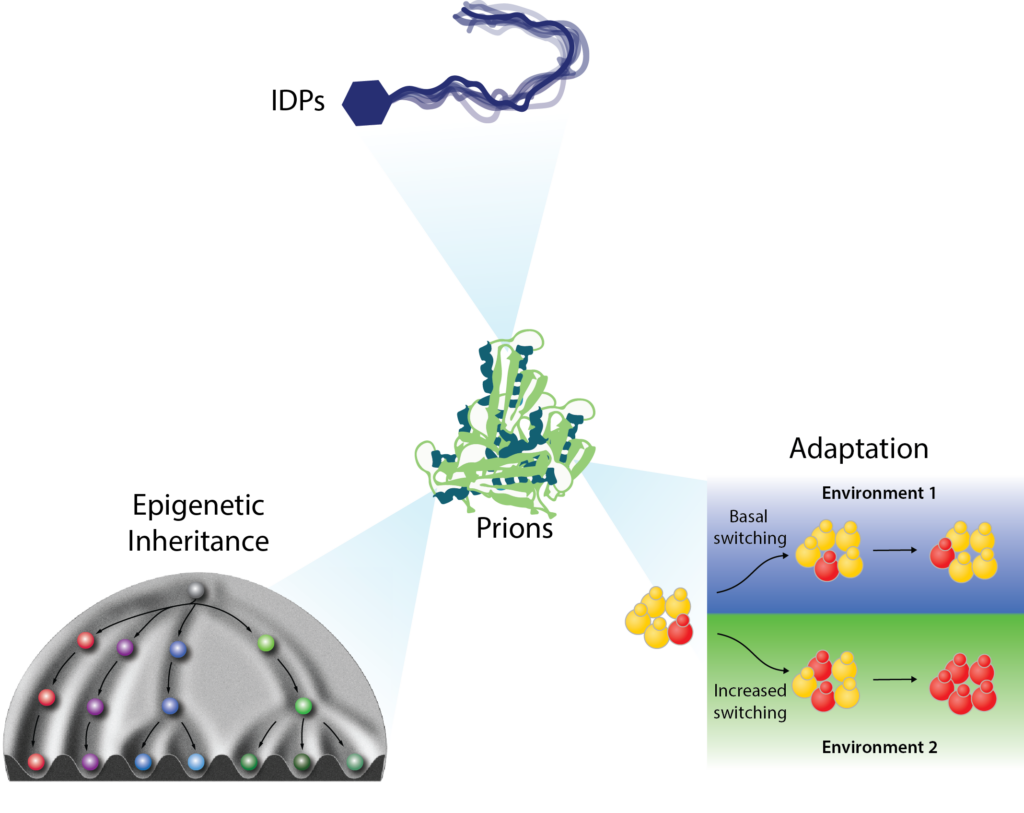
Control of growth and survival strategies – The protein-based epigenetic elements we have discovered have robust effects on physiology, for example controlling fundamental metabolic decisions, as for [GAR+]. Others fuel altered translational programs, such as [BIG+] (formed by the conserved pseudouridine synthase Pus4/TRUB1), which promotes enhanced growth and cell size at the expense of lifespan. [SMAUG+] provides a striking example of how pervasive such epigenetic elements can be. This prion governs the decision between two growth and survival strategies: whether to proliferate mitotically or differentiate into a stress resistant state via meiosis. It does this by downregulating coherent network of transcripts encoding proteins that interact to favor mitotic growth and repress meiosis. We have developed mathematical models suggesting that a heritable yet reversible epigenetic switch with these properties would provide advantages based on adaptive prediction in environments where starvation is regularly followed by nutrient replenishment. We found that laboratory yeast strains, which have experienced a regular pattern of nutrient depletion followed by replenishment, commonly harbor [SMAUG+]. It is also widespread in natural yeast isolates, which harbor distinct [SMAUG+] ‘polymorphs’ with differing assembly and hyperactivation properties, sparking heritable diversification in phenotype from the same protein sequence. These observations provide a powerful example of how a pervasive, protein-based form of epigenetic inheritance can be hidden in plain sight, profoundly altering growth and differentiation strategies. Given the widespread presence of the sequence features that drive this behavior in eukaryotic proteomes, we suspect that many more such examples remain to be discovered. In addition to the lines of investigation above we are examining the influence of this form of epigenetics on the acquisition of drug resistance in the human pathogen Candida albicans and in cancer cells.
↑ Back To Top

Finally, we are investigating protein aggregation during aging in yeast and, in collaboration with Anne Brunet (Stanford Genetics) in the African killifish. In yeast, alterations in maternal proteomes begin in middle age. Most are reset in daughters. In contrast, many changes persist in the daughters of old mothers, linked to the transmission of prion conformers of TFs and RBPs. Strikingly, de novo protein modification analyses point to site-specific N/Q oxidation as the most common damage to aging proteomes in yeast, Caenorhabditis elegans, and the African killifish, an emerging model for vertebrate aging. We are also harnessing the rapid aging process of African killifish, establishing a robust protocol to isolate aggregates from young and old animals. Mass spectrometry has identified many vertebrate-specific proteins that aggregate with age, often in a tissue-specific manner. Many are linked to age-related degenerative diseases and harbor prion-like domains. Taking advantage of the tractability of these multiple models, we are examining whether prion-like properties are a driving force in aging.

